Advanced Strategies for Food-Grade Protein Production: A New E. coli/Lactic Acid Bacteria Shuttle Vector for Improved Cloning and Food-Grade Expression



Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Plasmid Construction
2.3. Cassette Removal Tests
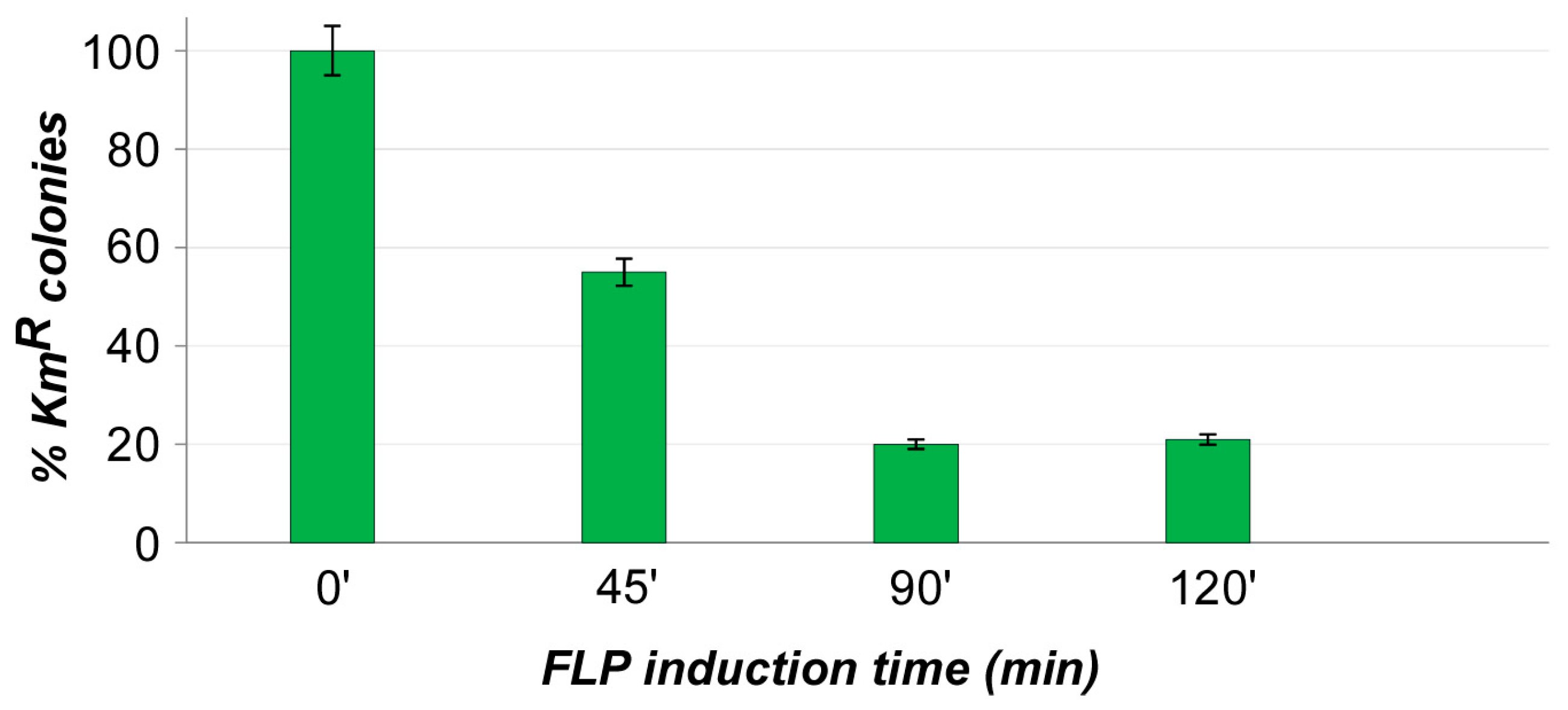
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rygus, T.; Hillen, W. Inducible high-level expression of heterologous genes in Bacillus megaterium using the regulatory elements of the xylose-utilization operon. Appl. Microbiol. Biotechnol. 1991, 35, 594–599. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Nguyen, Q.A.; Ferreira, R.C.; Ferreira, L.C.S.; Tran, L.T.; Schumann, W. Construction of plasmid-based expression vectors for Bacillus subtilis. Plasmid 2005, 54, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.T.P.; Nguyen, H.D.; Schumann, W. Novel plasmid-based expression vectors for intra- and extracellular production of recombinant proteins in Bacillus subtilis. Protein Expr. Purif. 2006, 46, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Malten, M.; Biedendieck, R.; Gamer, M.; Drews, A.-C.; Stammen, S.; Buchholz, K.; Dijkhuizen, L.; Jahn, D. A Bacillus megaterium plasmid system for the production, export and one-step purification of affinity tagged heterologous levansucrase from the growth medium. Appl. Environ. Microbiol. 2006, 72, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hollmann, R.; Deckwer, W.D. Comparative proteomic analysis of high cell density cultivations with two recombinant Bacillus megaterium strains for the production of a heterologous dextransucrase. Proteome Sci. 2006, 4, 19. [Google Scholar] [PubMed]
- Nguyen, H.D.; Phan, T.T.; Schumann, W. Expression Vectors for the Rapid Purification of Recombinant Proteins in Bacillus subtilis. Curr. Microbiol. 2007, 55, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Schumann, W. Production of recombinant proteins in Bacillus subtilis. Adv. Appl. Microbiol. 2007, 62, 137–189. [Google Scholar] [PubMed]
- Stammen, S.; Müller, B.K.; Korneli, C.; Biedendieck, R.; Gamer, M.; Franco-Lara, E.; Jahn, D. High-yield intra- and extracellular protein production using Bacillus megaterium. Appl. Microbiol. Biotechnol. 2010, 76, 4037–4046. [Google Scholar] [CrossRef]
- Kawabata, Y.; Kimura, K.; Funane, K. Extracellular production of cycloisomaltooligosaccharide glucanotransferase and cyclodextran by a protease-deficient Bacillus subtilis host-vector system. Appl. Microbiol. Biotechnol. 2012, 93, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Zhang, H.W.; Song, D.G.; Xie, Y.G.; Feng, J.; Wang, Y.Z. Expressing antimicrobial peptide cathelicidin-BF in Bacillus subtilis using SUMO technology. Appl. Microbiol. Biotechnol. 2014, 98, 3651–3658. [Google Scholar] [CrossRef]
- Zhang, K.; Su, L.; Duan, X.; Liu, L.; Wu, J. High-level extracellular protein production in Bacillus subtilis using an optimized dual-promoter expression system. Microb. Cell Fact. 2017, 16, 32. [Google Scholar] [CrossRef]
- Phan, T.; Huynh, P.; Truong, T.; Nguyen, H.A. Generic Protocol for Intracellular Expression of Recombinant Proteins in Bacillus subtilis. Methods Mol. Biol. 2017, 1586, 325–334. [Google Scholar]
- Landete, J.M. A review of food-grade vectors in lactic acid bacteria: From the laboratory to their application. Crit. Rev. Biotechnol. 2017, 37, 296–308. [Google Scholar] [CrossRef]
- van de Guchte, M.; Penaud, S.; Grimaldi, C.; Barbe, V.; Bryson, K.; Nicolas, P.; Robert, C.; Oztas, S.; Mangenot, S.; Couloux, A.; et al. The complete genome sequence of Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 9274–9279. [Google Scholar] [CrossRef]
- Wells, J. Mucosal vaccination and therapy with genetically modified lactic acid bacteria. Annu. Rev. Food Sci. Technol. 2011, 2, 423–445. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.B.; Saraiva, T.D.L.; Souza, B.M.; Zurita-Turk, M.; Azevedo, M.S.P.; De Castro, C.P.; Mancha-Agresti, P.; dos Santos, J.S.C.; Santos, A.C.G.; Faria, A.M.C.; et al. Development of a new DNA vaccine based on mycobacterial ESAT-6 antigen delivered by recombinant invasive Lactococcus lactis FnBPA+. Appl. Microbiol. Biotechnol. 2015, 99, 1817–1826. [Google Scholar] [CrossRef]
- Pereira, V.B.; da Cunha, V.P.; Preisser, T.M.; Souza, B.M.; Turk, M.Z.; De Castro, C.P.; Azevedo, M.S.P.; Miyoshi, A. Lactococcus lactis carrying a DNA vaccine coding for the ESAT-6 antigen increases IL-17 cytokine secretion and boosts the BCG vaccine immune response. J. Appl. Microbiol. 2017, 122, 1657–1662. [Google Scholar] [CrossRef]
- Pontes, D.; Azevedo, M.; Innocentin, S.; Blugeon, S.; Lefévre, F.; Azevedo, V.; Miyoshi, A.; Courtin, P.; Chapot-Chartier, M.-P.; Langella, P.; et al. Immune response elicited by DNA vaccination using Lactococcus lactis is modified by the production of surface exposed pathogenic protein. PLoS ONE 2014, 9, e84509. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Turk, M.; Del Carmen, S.; Santos, A.C.G.; Pereira, V.B.; Cara, D.C.; Leclercq, S.Y.; de LeBlanc, A.D.; Azevedo, V.; Chatel, J.-M.; LeBlanc, J.G.; et al. Lactococcus lactis carrying the pValac DNA expression vector coding for IL-10 reduces inflammation in a murine model of experimental colitis. BMC Biotechnol. 2014, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Mancha-Agresti, P.; de Castro, C.P.; Dos Santos, J.S.C.; Araujo, M.A.; Pereira, V.B.; LeBlanc, J.G.; Leclercq, S.Y.; Azevedo, V. Recombinant Invasive Lactococcus lactis Carrying a DNA Vaccine Coding the Ag85A Antigen Increases INF-γ, IL-6, and TNF-α Cytokines after Intranasal Immunization. Front. Microbiol. 2017, 8, 1263. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.M.; Preisser, T.M.; Pereira, V.B.; Zurita-Turk, M.; de Castro, C.P.; da Cunha, V.P.; de Oliveira, R.P.; Gomes-Santos, A.C.; de Faria, A.M.C.; Machado, D.C.C.; et al. Lactococcus lactis carrying the pValac eukaryotic expression vector coding for IL-4 reduces chemically-induced intestinal inflammation by increasing the levels of IL-10-producing regulatory cells. Microb. Cell Fact. 2016, 15, 150. [Google Scholar] [CrossRef]
- Mercenier, A.; Müller-Alouf, H.; Grangette, C. Lactic acid bacteria as live vaccines. Curr. Issues Mol. Biol. 2000, 2, 17–25. [Google Scholar]
- Pontes, D.S.; de Azevedo, M.S.P.; Chatel, J.-M.; Langella, P.; Azevedo, V.; Miyoshi, A. Lactococcus lactis as a live vector: Heterologous protein production and DNA delivery systems. Protein Expr. Purif. 2011, 79, 165–175. [Google Scholar] [CrossRef]
- Kok, J.; van der Vossen, J.M.; Venema, G. Construction of plasmid cloning vectors for lactic streptococci which also replicate in Bacillus subtilis and Escherichia coli. Appl. Environ. Microbiol. 1984, 48, 726–731. [Google Scholar] [PubMed]
- de Vos, W.M.; Kleerebezem, M.; Kuipers, O.P. Expression systems for industrial Gram-positive bacteria with low guanine and cytosine content. Curr. Opin. Biotechnol. 1997, 8, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Benbouziane, B.; Ribelles, P.; Aubry, C.; Martin, R.; Kharrat, P.; Riazi, A.; Langella, P.; Bermúdez-Humarán, L.G. Development of a Stress-Inducible Controlled Expression (SICE) system in Lactococcus lactis for the production and delivery of therapeutic molecules at mucosal surfaces. J. Biotechnol. 2013, 168, 120–129. [Google Scholar] [CrossRef]
- Mu, D.; Montalbán-López, M.; Masuda, Y.; Kuipers, O.P. Zirex: A Novel Zinc-Regulated Expression System for Lactococcus lactis. Appl. Environ. Microbiol. 2013, 79, 4503–4508. [Google Scholar] [CrossRef] [PubMed]
- de Castro, C.P.; Drumond, M.M.; Batista, V.L.; Nunes, A.; Mancha-Agresti, P.; Azevedo, V. Vector Development Timeline for Mucosal Vaccination and Treatment of Disease Using Lactococcus lactis and Design Approaches of Next Generation Food Grade Plasmids. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Yin, S.; Zhu, H.; Shen, M.; Li, G.; Lu, S.; Zhao, Y.; Le, S.; Tan, Y.; Peng, Y.; Hu, F.; et al. Surface Display of Heterologous β-Galactosidase in Food-Grade Recombinant Lactococcus lactis. Curr. Microbiol. 2018, 75, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C.; Ross, R.P. A food-grade approach for functional analysis and modification of native plasmids in Lactococcus lactis. Appl. Environ. Microbiol. 2003, 69, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Shin, J.; Cho, B.-K. Applications of CRISPR/Cas System to Bacterial Metabolic Engineering. Int. J. Mol. Sci. 2018, 19, 1089. [Google Scholar] [CrossRef]
- Berlec, A.; Strukelj, B. Novel applications of recombinant lactic acid bacteria in therapy and in metabolic engineering. Recent Pat. Biotechnol. 2009, 3, 77–87. [Google Scholar] [CrossRef]
- Berlec, A.; Škrlec, K.; Kocjan, J.; Olenic, M.; Štrukelj, B. Single plasmid systems for inducible dual protein expression and for CRISPR-Cas9/CRISPRi gene regulation in lactic acid bacterium Lactococcus lactis. Sci. Rep. 2018, 8, 1009. [Google Scholar] [CrossRef]
- Gosalbes, M.J.; Esteban, C.D.; Galán, J.L.; Pérez-Martínez, G. Integrative Food-Grade Expression System Based on the Lactose Regulon of Lactobacillus casei. Appl. Environ. Microbiol. 2000, 66, 4822–4828. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Ling, P.; Wang, F. Constructing a recombinant hyaluronic acid biosynthesis operon and producing food-grade hyaluronic acid in Lactococcus lactis. J. Ind. Microbiol. Biotechnol. 2015, 42, 197–206. [Google Scholar] [CrossRef]
- MacCormick, C.A.; Griffin, H.G.; Gasson, M.J. Construction of a food-grade host/vector system for Lactococcus lactis based on the lactose operon. FEMS Microbiol. Lett. 1995, 127, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Platteeuw, C.; van Alen-Boerrigter, I.; van Schalkwijk, S.; de Vos, W.M. Food-grade cloning and expression system for Lactococcus lactis. Appl. Environ. Microbiol. 1996, 62, 1008–1013. [Google Scholar]
- de Ruyter, P.G.; Kuipers, O.P.; de Vos, W.M. Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl. Environ. Microbiol. 1996, 62, 3662–3667. [Google Scholar] [PubMed]
- Gu, P.; Yang, F.; Su, T.; Wang, Q.; Liang, Q.; Qi, Q. A rapid and reliable strategy for chromosomal integration of gene(s) with multiple copies. Sci. Rep. 2015, 5, 9684. [Google Scholar] [CrossRef]
- Ou, B.; Garcia, C.; Wang, Y.; Zhang, W.; Zhu, G. Techniques for chromosomal integration and expression optimization in Escherichia coli. Biotechnol. Bioeng. 2018, 115, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-F.; Sun, M.-Y.; Cui, L.-Y.; Zhang, C.; Xing, X.-H. Cre/loxP-Mediated Multicopy Integration of the Mevalonate Operon into the Genome of Methylobacterium extorquens AM1. Appl. Biochem. Biotechnol. 2018, 185, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M. The FLP protein of the yeast 2-microns plasmid: Expression of a eukaryotic genetic recombination system in Escherichia coli. Proc. Natl. Acad. Sci. USA 1983, 80, 4223–4227. [Google Scholar] [CrossRef] [PubMed]
- Gronostajski, R.M.; Sadowski, P.D. The FLP recombinase of the Saccharomyces cerevisiae 2 microns plasmid attaches covalently to DNA via a phosphotyrosyl linkage. Mol. Cell. Biol. 1985, 5, 3274–3279. [Google Scholar] [CrossRef]
- Senecoff, J.F.; Bruckner, R.C.; Cox, M.M. The FLP recombinase of the yeast 2-micron plasmid: Characterization of its recombination site. Proc. Natl. Acad. Sci. USA 1985, 82, 7270–7274. [Google Scholar] [CrossRef]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Jensen, S.I.; Lennen, R.M.; Herrgård, M.J.; Nielsen, A.T. Seven gene deletions in seven days: Fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Sci. Rep. 2015, 5, 17874. [Google Scholar] [CrossRef] [PubMed]
- Tagliavia, M.; Nicosia, A.; Salamone, M.; Biondo, G.; Bennici, C.D.; Mazzola, S.; Cuttitta, A. Development of a fast DNA extraction method for sea food and marine species identification. Food Chem. 2016, 203, 375–378. [Google Scholar] [CrossRef]
- Mendez-Perez, D.; Gunasekaran, S.; Orler, V.J.; Pfleger, B.F. A translation-coupling DNA cassette for monitoring protein translation in Escherichia coli. Metab. Eng. 2012, 14, 298–305. [Google Scholar] [CrossRef]
- Tagliavia, M.; Cuttitta, A. Exploiting translational coupling for the selection of cells producing toxic recombinant proteins from expression vectors. BioTechniques 2016, 60, 113–118. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Primer Name | Sequence (5′-3′) |
|---|---|
| pNZ2122_680F | CAGGTACCACTAGTTTAGTTCCACGTGGTAGTCATCATCATCATCATCATTAATTTATAAATAAAAATCACCTTTTAGAG |
| pNZ2122_395R | AGTGGTACCTGGATCCACTCGAGGTCGACGAGCTCTTGAATTCATTTGGACTACCTCCTAAAT |
| pNZ2122/803ClaF | TCAAATCGATTCCACCAATTAAAGGACCGATAAC |
| pNZ2122/760SpeR | TCAACTAGTATTCTGCTCCCGCCCTTATG |
| pKD13_P1Cla | TCAAATCGATGTGTAGGCTGGAGCTGCTTC |
| pKD13_P4Spe | TCAACTAGTGAATTAATTCCGGAGATCCATCGACGTGCAGTTC |
| pKD13_P1 | GTGTAGGCTGGAGCTGCTTC |
| pKD13_P41 | GAGATCCATCGACGTGCAGTTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tagliavia, M.; Nicosia, A. Advanced Strategies for Food-Grade Protein Production: A New E. coli/Lactic Acid Bacteria Shuttle Vector for Improved Cloning and Food-Grade Expression. Microorganisms 2019, 7, 116. https://doi.org/10.3390/microorganisms7050116
Tagliavia M, Nicosia A. Advanced Strategies for Food-Grade Protein Production: A New E. coli/Lactic Acid Bacteria Shuttle Vector for Improved Cloning and Food-Grade Expression. Microorganisms. 2019; 7(5):116. https://doi.org/10.3390/microorganisms7050116
Chicago/Turabian StyleTagliavia, Marcello, and Aldo Nicosia. 2019. "Advanced Strategies for Food-Grade Protein Production: A New E. coli/Lactic Acid Bacteria Shuttle Vector for Improved Cloning and Food-Grade Expression" Microorganisms 7, no. 5: 116. https://doi.org/10.3390/microorganisms7050116
APA StyleTagliavia, M., & Nicosia, A. (2019). Advanced Strategies for Food-Grade Protein Production: A New E. coli/Lactic Acid Bacteria Shuttle Vector for Improved Cloning and Food-Grade Expression. Microorganisms, 7(5), 116. https://doi.org/10.3390/microorganisms7050116