A New Genome-to-Genome Comparison Approach for Large-Scale Revisiting of Current Microbial Taxonomy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Description
2.2. Homologous Coverage Ratio (HCR)
2.3. ANI and Other Tools
3. Results
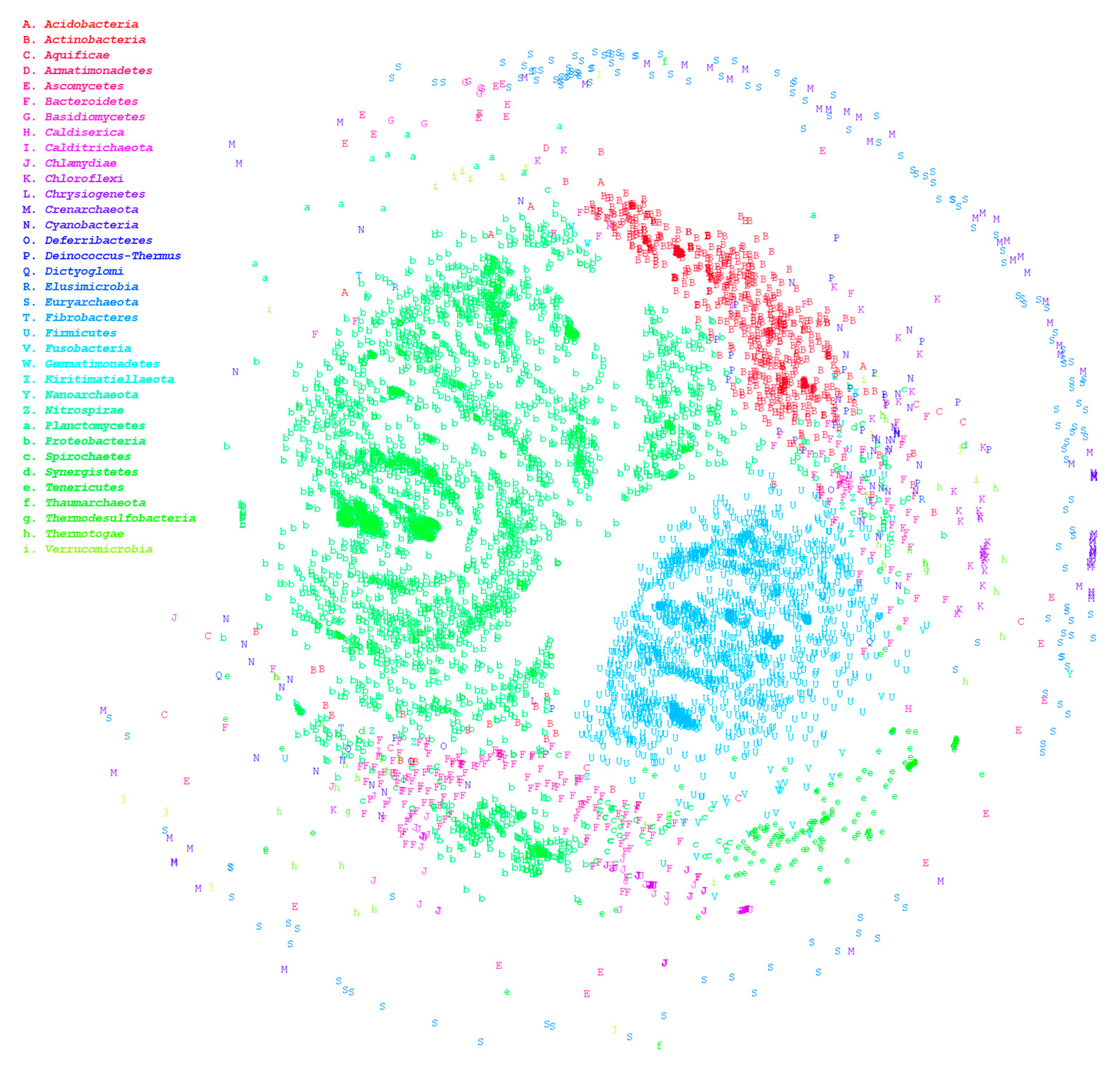
3.1. Large-Scale Genome Comparison
3.2. Comparison of HCR and ANI Methods
3.3. Validation by BAC120
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pang, T.Y.; Lercher, M. The adaptive acquisition of single DNA segments drives metabolic evolution across E. coli lineages. Arxiv Prepr. 2015, 1509, 06667. [Google Scholar]
- Vos, M.; Hesselman, M.C.; Te Beek, T.A.; van Passel, M.W.J.; Eyre-Walker, A. Rates of Lateral Gene Transfer in Prokaryotes: High but Why? Trends Microbiol. 2015, 23, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Marcelino, L.A.; Klepac-Ceraj, V.; Polz, M.F. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 2004, 186, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Rainey, F.A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Anton Leeuw Int. J. G 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Galperin, M.Y. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 2013, 15, 2631–2641. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Phylum | Minimum Homologous Coverage Ratio of Intragenus | Assembly Accession | Assembly Accession |
|---|---|---|---|
| Proteobacteria | 0.015999617 | GCA_001417865.2 | GCA_001190755.1 |
| Tenericutes | 0.024333677 | GCA_000186985.3 | GCA_001886855.1 |
| Firmicutes | 0.026170854 | GCA_900183405.1 | GCA_001010825.1 |
| Spirochaetes | 0.048420481 | GCA_001936255.1 | GCA_000092845.1 |
| Fusobacteria | 0.140362380 | GCA_002356455.1 | GCA_000024565.1 |
| Bacteroidetes | 0.166804691 | GCA_000348805.1 | GCA_002369955.1 |
| Cyanobacteria | 0.216424948 | GCA_000015705.1 | GCA_000015665.1 |
| Thermotogae | 0.220607538 | GCA_000953715.1 | GCA_001941385.1 |
| Aquificae | 0.221942947 | GCA_000021545.1 | GCA_000191045.1 |
| Dictyoglomi | 0.749914757 | GCA_000020965.1 | GCA_000021645.1 |
| Phylum A | Phylum B | Minimum Homologous Coverage Ratio of Intragenus (Phylum A) | Maximum Homologous Coverage Ratio of intergenus | Assembly Accession | Assembly Accession |
|---|---|---|---|---|---|
| Proteobacteria | Bacteroidetes | 0.015999617 | 0.151368772 | GCA_002355135.1 | GCA_000090965.1 |
| Proteobacteria | Deferribacteres | 0.015999617 | 0.112992155 | GCA_000284355.1 | GCA_000010985.1 |
| Proteobacteria | Aquificae | 0.015999617 | 0.112416204 | GCA_000284355.1 | GCA_000021545.1 |
| Firmicutes | Dictyoglomi | 0.026170854 | 0.170361824 | GCA_000092965.1 | GCA_000020965.1 |
| Proteobacteria | Tenericutes | 0.015999617 | 0.101309783 | GCA_001262715.1 | GCA_900016775.1 |
| Proteobacteria | Firmicutes | 0.015999617 | 0.096263109 | GCA_000284355.1 | GCA_000014125.1 |
| Proteobacteria | Fusobacteria | 0.015999617 | 0.093542786 | GCA_000816185.1 | GCA_001296125.1 |
| Firmicutes | Deferribacteres | 0.026170854 | 0.13067773 | GCA_000165465.1 | GCA_000010985.1 |
| Proteobacteria | Cyanobacteria | 0.015999617 | 0.077366777 | GCA_000011465.1 | GCA_000008885.1 |
| Proteobacteria | Dictyoglomi | 0.015999617 | 0.074348485 | GCA_002220775.1 | GCA_000021645.1 |
| Firmicutes | Aquificae | 0.026170854 | 0.119079275 | GCA_000025645.1 | GCA_000191045.1 |
| Tenericutes | Firmicutes | 0.024333677 | 0.106030272 | GCA_001702115.1 | GCA_002441935.1 |
| Tenericutes | Fusobacteria | 0.024333677 | 0.104966983 | GCA_000439435.1 | GCA_000024565.1 |
| Genus A | Genus B | Max HCR of Cross-Genus | Genome Comparison of Cross-Genus | Max HCR of Intragenus | Genome Comparison of Intragenus |
|---|---|---|---|---|---|
| Alicycliphilus | Acidovorax | 0.521 | GCF_000175235.1 & GCF_000204645.1 | 0.6 | GCF_000175235.1 & GCF_002157165.1 |
| Chlorobium | Chlorobaculum | 0.324 | GCF_000012585.1 & GCF_000006985.1 | 0.37 | GCF_000012585.1 & GCF_000020645.1 |
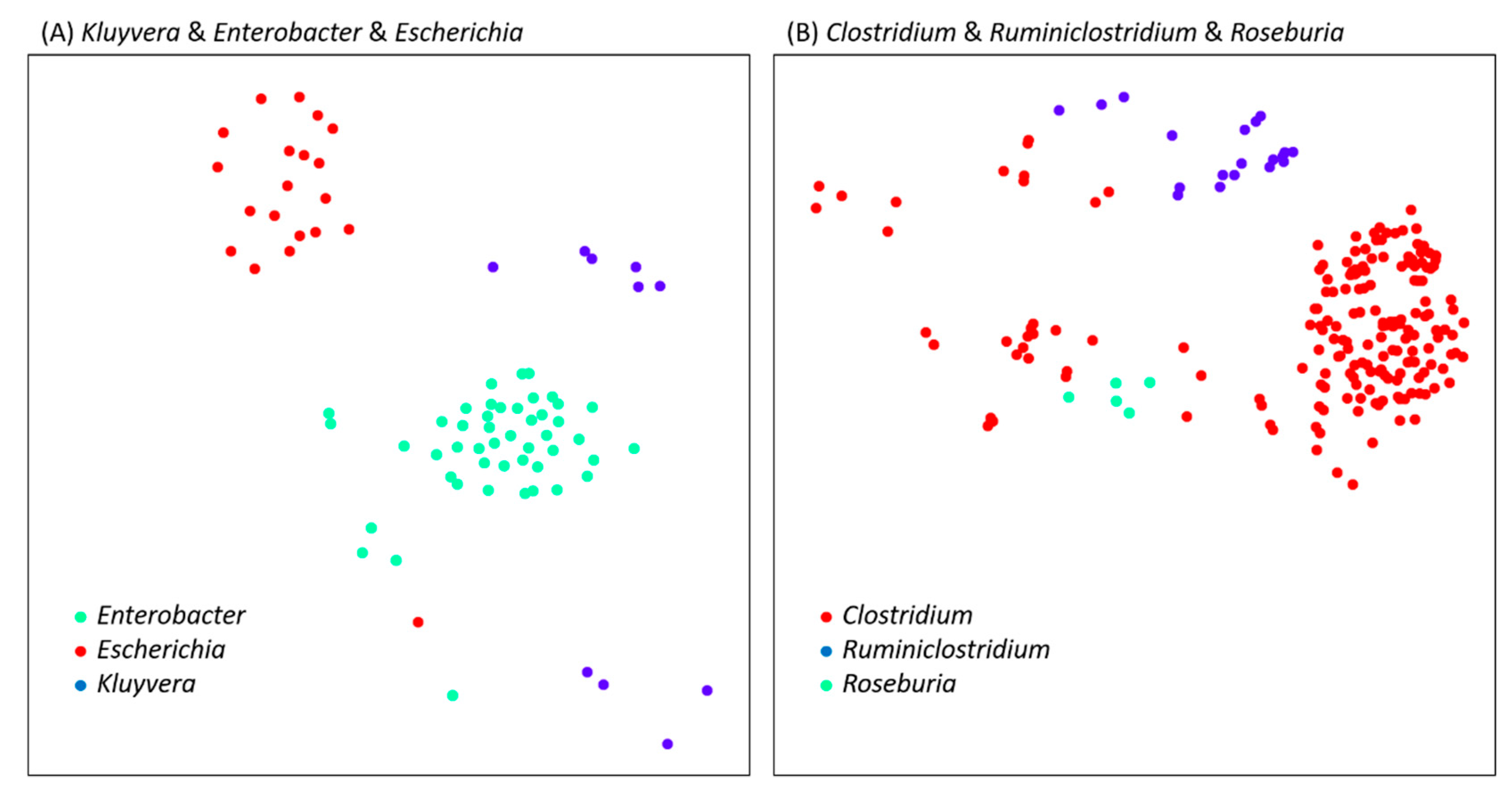
| Clostridium | Ruminiclostridium | 0.192 | GCF_000620945.1 & GCF_002161175.1 | 0.121 | GCF_000620945.1 & GCF_000953215.1 |
| Corynebacterium | Brevibacterium | 0.228 | GCF_000720035.1 & GCF_900184225.1 | 0.19 | GCF_000720035.1 & GCF_001941505.1 |
| Diaphorobacter | Acidovorax | 0.515 | GCF_000175235.1 & GCF_000015545.1 | 0.6 | GCF_000175235.1 & GCF_002157165.1 |
| Erythrobacter | Altererythrobacter | 0.479 | GCF_000013005.1 & GCF_900177715.1 | 0.493 | GCF_000013005.1 & GCF_900115585.1 |
| Histophilus | Haemophilus | 0.481 | GCF_002015075.1 & GCF_000027305.1 | 0.557 | GCF_002015075.1 & GCF_000011785.1 |
| Kluyvera | Enterobacter | 0.598 | GCF_000321045.1 & GCF_900168315.1 | 0.578 | GCF_000321045.1 & GCF_001888805.2 |
| Kluyvera | Escherichia | 0.549 | GCF_000759795.1 & GCF_900112785.1 | 0.506 | GCF_000759795.1 & GCF_000350705.1 |
| Lelliottia | Enterobacter | 0.709 | GCF_001652505.2 & GCF_001729725.1 | 0.702 | GCF_001652505.2 & GCF_002811785.1 |
| Pseudodesulfovibrio | Desulfovibrio | 0.397 | GCF_000422565.1 & GCF_000189295.2 | 0.397 | GCF_000422565.1 & GCF_900188225.1 |
| Roseburia | Clostridium | 0.194 | GCF_900111235.1 & GCF_001940165.1 | 0.2 | GCF_900111235.1 & GCF_900112775.1 |
| Serratia | Chania | 0.575 | GCF_001976145.1 & GCF_002588845.1 | 0.697 | GCF_001976145.1 & GCF_000743365.1 |
| Sphingomonas | Rhizorhabdus | 0.33 | GCF_000512205.2 & GCF_000715175.2 | 0.362 | GCF_000512205.2 & GCF_001717955.1 |
| Vibrio | Aliivibrio | 0.312 | GCF_002100145.1 & GCF_001691025.1 | 0.364 | GCF_002100145.1 & GCF_000280885.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-H.; Liu, Y.-Y.; Soo, V.-W.; Chen, C.-C. A New Genome-to-Genome Comparison Approach for Large-Scale Revisiting of Current Microbial Taxonomy. Microorganisms 2019, 7, 161. https://doi.org/10.3390/microorganisms7060161
Tsai M-H, Liu Y-Y, Soo V-W, Chen C-C. A New Genome-to-Genome Comparison Approach for Large-Scale Revisiting of Current Microbial Taxonomy. Microorganisms. 2019; 7(6):161. https://doi.org/10.3390/microorganisms7060161
Chicago/Turabian StyleTsai, Ming-Hsin, Yen-Yi Liu, Von-Wun Soo, and Chih-Chieh Chen. 2019. "A New Genome-to-Genome Comparison Approach for Large-Scale Revisiting of Current Microbial Taxonomy" Microorganisms 7, no. 6: 161. https://doi.org/10.3390/microorganisms7060161
APA StyleTsai, M.-H., Liu, Y.-Y., Soo, V.-W., & Chen, C.-C. (2019). A New Genome-to-Genome Comparison Approach for Large-Scale Revisiting of Current Microbial Taxonomy. Microorganisms, 7(6), 161. https://doi.org/10.3390/microorganisms7060161