Diversity and Dynamics of Seaweed Associated Microbial Communities Inhabiting the Lagoon of Venice

 ,
, 


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Water Sampling and Environmental Parameters Monitoring
2.3. Microbial Community Analyses
2.3.1. Seaweed Sampling
2.3.2. DNA Extraction, Sequencing and 16S rRNA Sequence Data Processing
2.4. Microbial Community Structure
2.5. Linear Discriminant Analysis (LDA) Effect Size (LEfSe) Measurement
2.6. Relationship between SAMCs and Environmental Parameters
3. Results
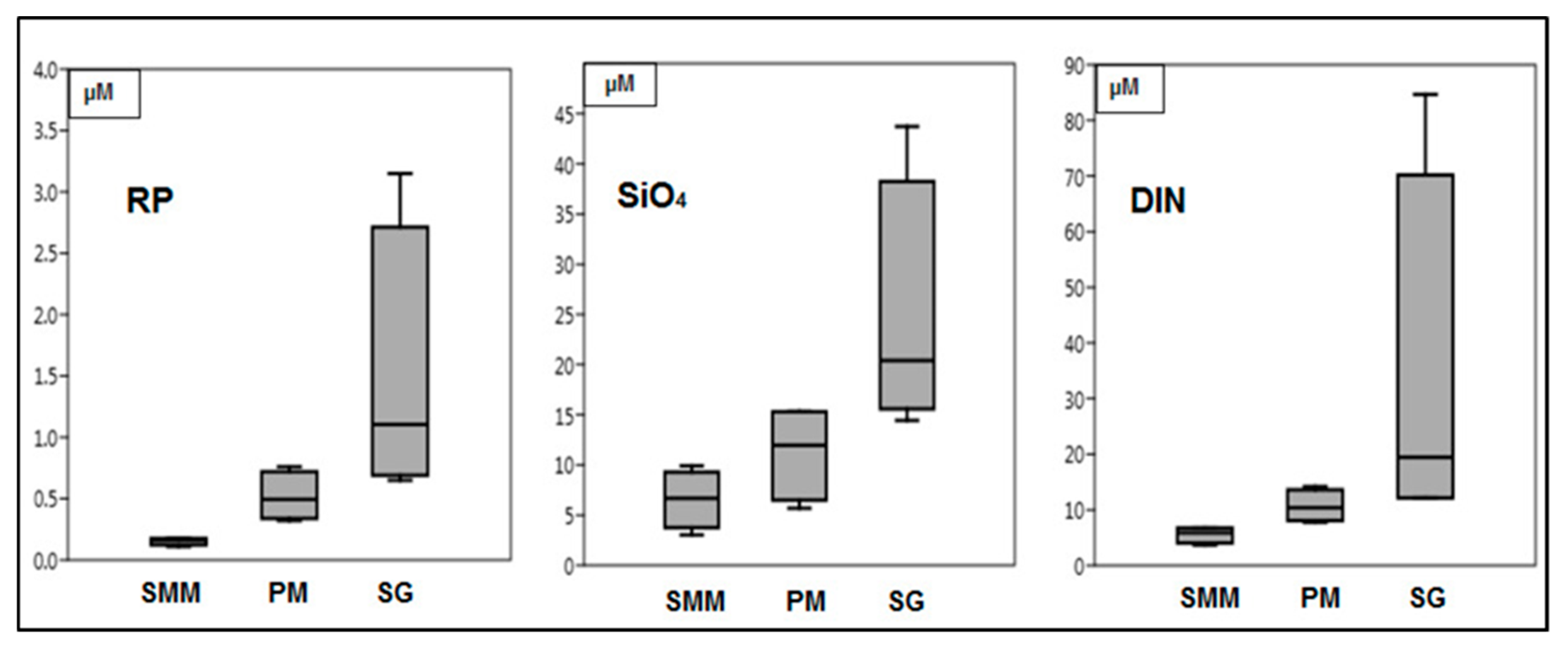
3.1. Environmental Parameters Analyses
3.2. Overview of Sequencing Output
3.3. Microbial Communities’ Structure
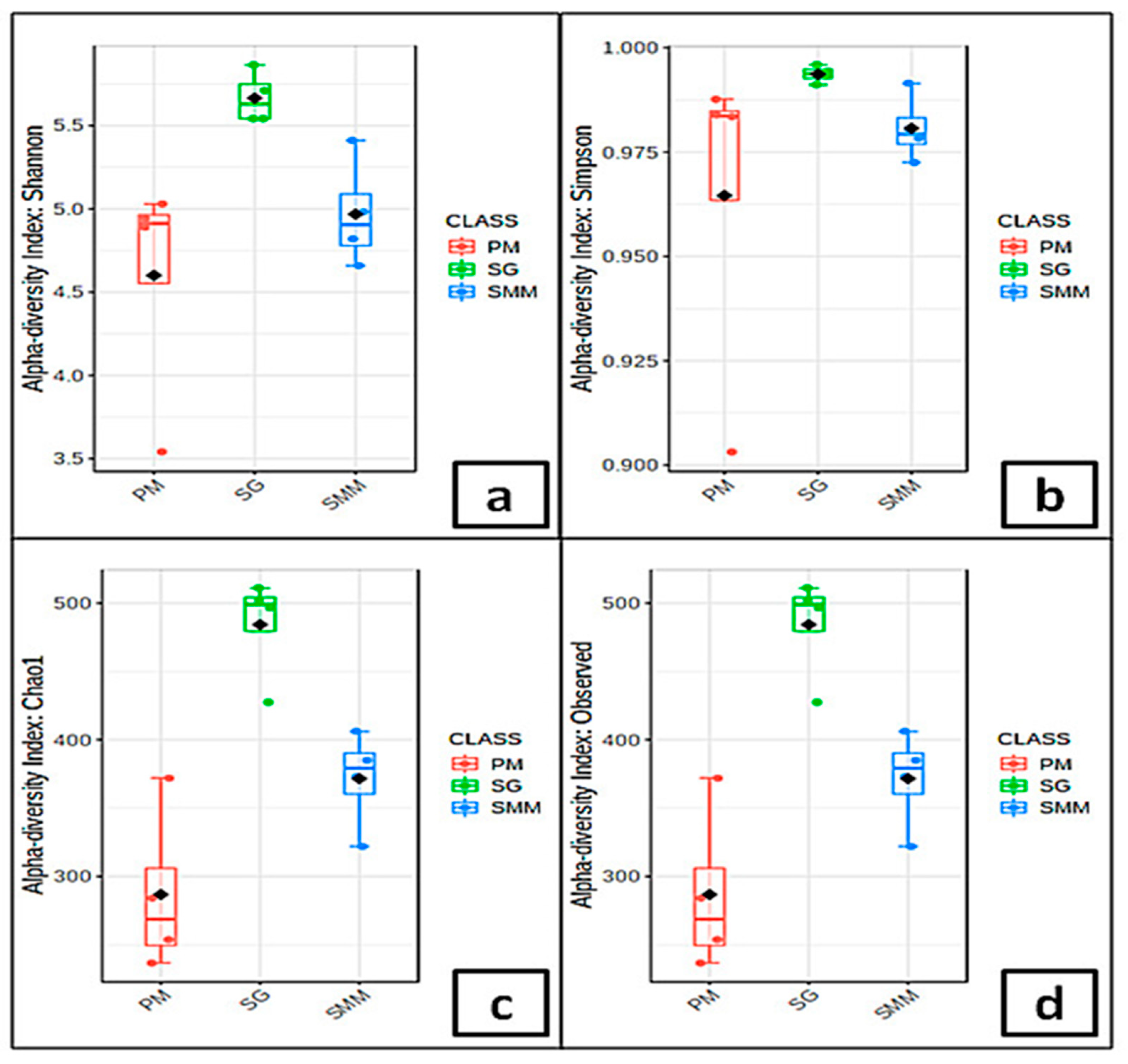
3.4. Microbial Communities’ Indices
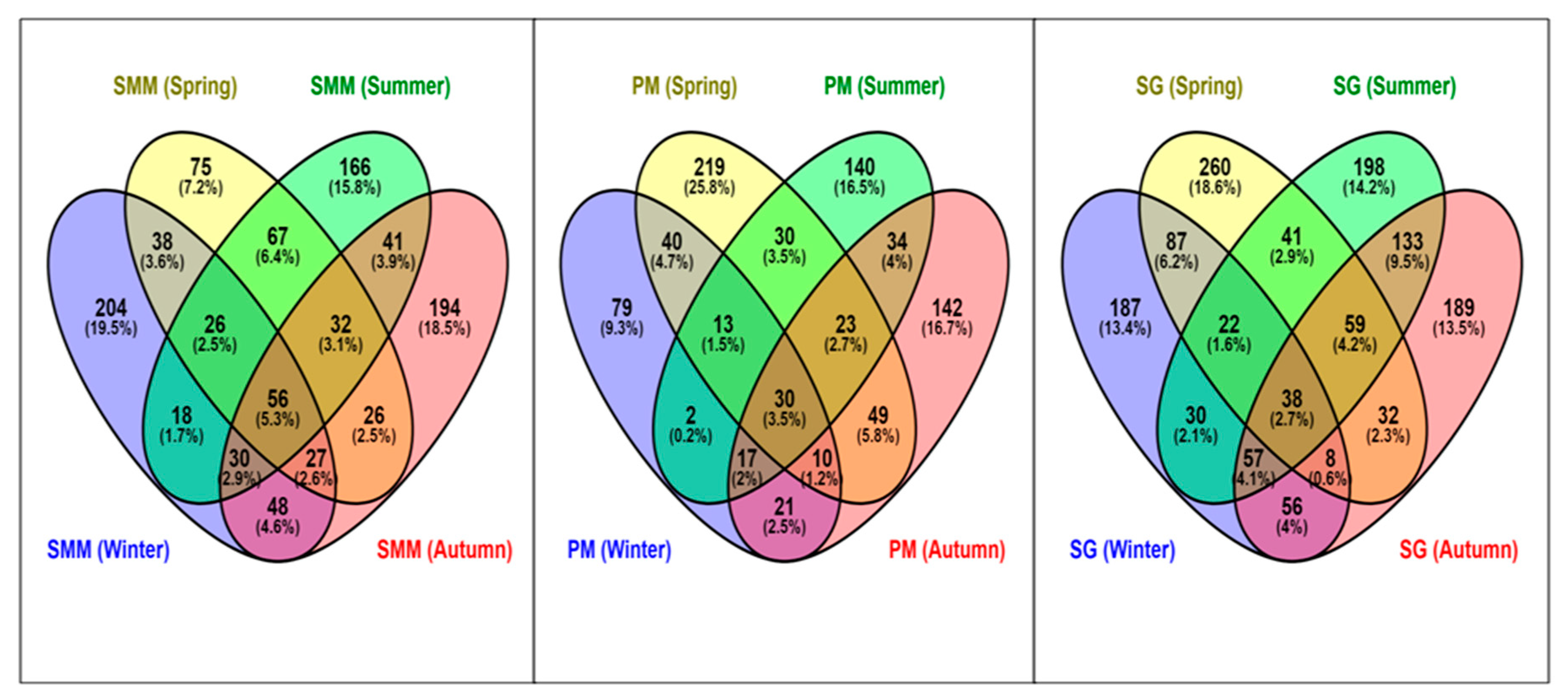
3.5. Distribution of Shared and Non-Ubiquitous ASVs of SAMCs
3.6. Linear Discriminant Analysis (LDA) Effect Size (LEfSe) Characterization of SAMCs
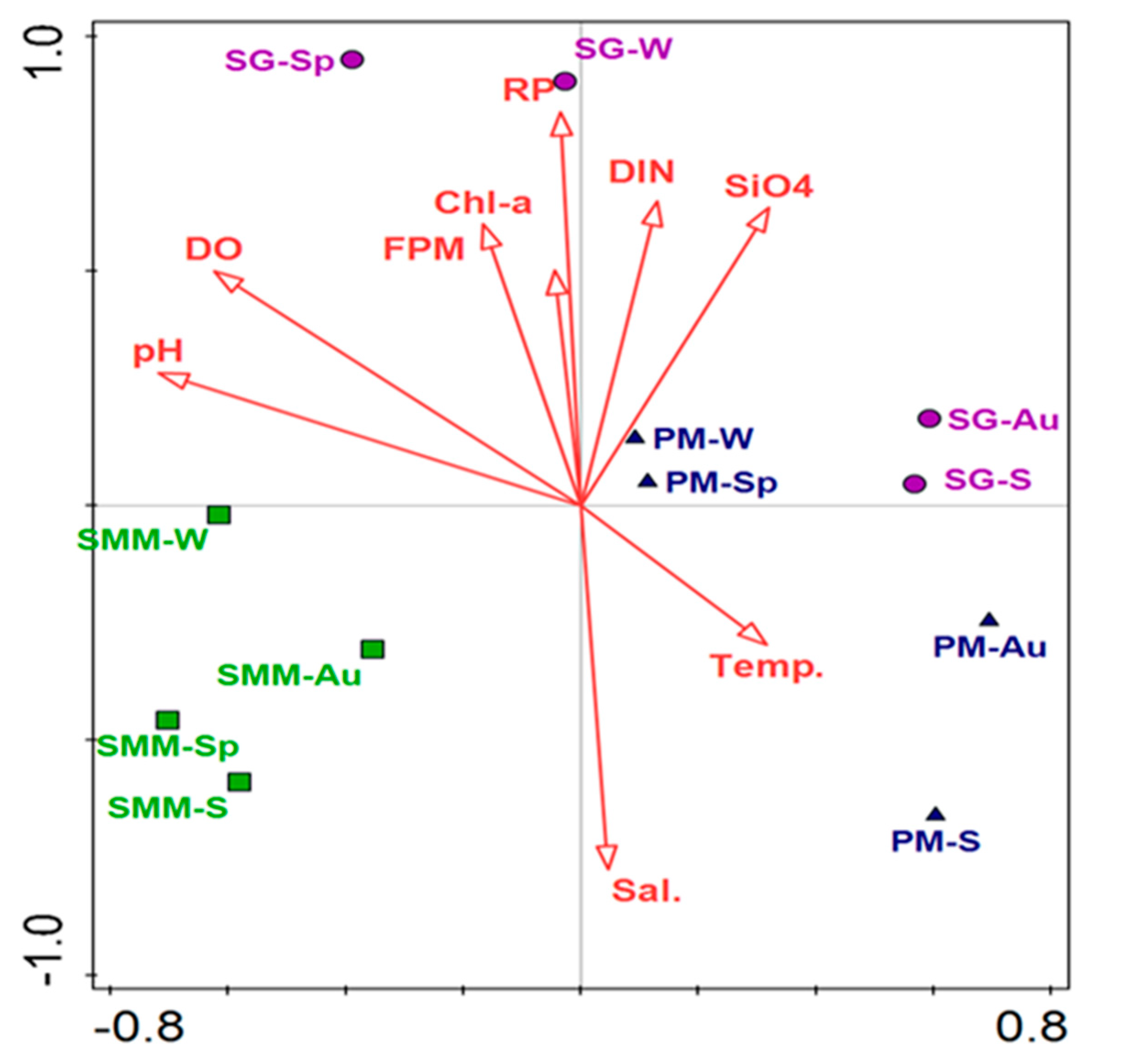
3.7. Correlation between Bacterial Communities and Environmental Parameters
4. Discussion
4.1. Characterization of Microbial Communities
4.2. Seasonal Variations of SAMCs
4.3. Microbial Communities and Environmental Stressors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, P.D.; de-Nys, R.; Kjelleberg, S. Chemical cues for surface colonization. J. Chem. Ecol. 2002, 28, 1935–1951. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Reddy, C. Seaweed-microbial interactions: Key functions of seaweed-associated bacteria. Fems Microbiol. Ecol. 2014, 88, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.M.; Sjøtun, K.; Lanzén, A.; Øvreås, L. Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborean. Isme J. 2012, 6, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; Sharon, G.; Atad, I.; Zilber-Rosenberg, I. The evolution of animals and plants via symbiosis with microorganisms. Environ. Microbiol. Rep. 2010, 2, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Aires, T.; Serrão, E.A.; Kendrick, G.; Duarte, C.M.; Arnaud-Haond, S. Invasion Is a Community Affair: Clandestine Followers in the Bacterial Community Associated to Green Algae Caulerpa racemosa, Track the Invasion Source. PLoS ONE 2013, 8, e68429. [Google Scholar] [CrossRef] [Green Version]
- Aires, T.; Moalic, Y.; Serrao, E.A.; Arnaud-Haond, S. Hologenome theory supported by co-occurrence networks of species-specific bacterial communities in Siphonous algae (Caulerpa). Fems Microbiol. Ecol. 2015, 91, fiv067. [Google Scholar] [CrossRef] [Green Version]
- Head, I.M.; Jones, D.M.; Röling, W.F.M. Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 2006, 4, 173–182. [Google Scholar] [CrossRef]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed–bacteria interactions. Fems Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Goecke, F.; Thiel, V.; Wiese, J.; Labes, A.; Imhoff, J.F. Algae as an important environment for bacteria – phylogenetic relationships among new bacterial species isolated from algae. Phycologia 2013, 52, 14–24. [Google Scholar] [CrossRef]
- Marzinelli, E.M.; Campbell, A.H.; Zozaya Valdes, E.; Vergés, A.; Nielsen, S.; Wernberg, T.; Bettignies, T.; Bennett, S.; Caporaso, J.G.; Thomas, T.; et al. Host condition explains bacterial communities. Environ. Microbiol. 2015, 17, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Michelou, V.K.; Caporaso, J.G.; Knight, R.; Palumbi, S.R. The ecology of microbial communities associated with Macrocystis p.yrifera. PLoS ONE 2013, 8, e67480. [Google Scholar] [CrossRef]
- Wahl, M.; Molis, M.; Hobday, A.J.; Dudgeon, S.R.; Neumann, R.; Steinberg, P.; Campbell, A.H.; Marzinelli, E.; Connell, S.D. The responses of brown macroalgae to environmental change from local to global scales: Direct and ecologically mediated effects. Perspect. Phycol. 2015, 2, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Lachnit, T.; Blümel, M.; Imhoff, J.; Wahl, M. Specific epibacterial communities on macroalgae: Phylogeny matters more than habitat. Aquat Biol. 2009, 5, 181–186. [Google Scholar] [CrossRef]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 2011, 13, 655–665. [Google Scholar] [CrossRef]
- Marzinelli, E.M.; Qiu, Z.; Dafforn, K.A.; Johnston, E.L.; Steinberg, P.D.; Mayer-Pinto, M. Coastal urbanisation affects microbial communities on a dominant marine holobiont. Npj Biofilms Microbiomes 2018, 4, 1–7. [Google Scholar] [CrossRef]
- Aires, T.; Serrão, E.; Engelen, A. Host and Environmental Specificity in Bacterial Communities Associated to Two Highly Invasive Marine Species (Genus Asparagopsis). Front. Microbiol. 2016, 7, 559. [Google Scholar] [CrossRef]
- Sfriso, A. Coexistence of Ulva rigida and Ulva laetevirens (Ulvales, Chlorophyta) in Venice Lagoon and other Italian transitional and marine environments. Bot. Mar. 2010, 53, 9–18. [Google Scholar] [CrossRef]
- Sfriso, A.; Buosi, A.; Mistri, M.; Munari, C.; Franzoi, P.; Sfriso, A.A. Long-term changes of the trophic status in transitional ecosystems of the northern Adriatic Sea, key parameters and future expectations: The lagoon of Venice as a study case. In Italian Long-Term Ecological Research for Understanding Ecosystem Diversity and Functioning. Case Studies from Aquatic, Terrestrial and Transitional Domains; Mazzocchi, M.G., Capotondi, L., Freppaz, M., Lugliè, A., Campanaro, A., Eds.; Nature Conservation: Bulgaria, 2019; pp. 193–215. [Google Scholar] [CrossRef]
- Facca, C.; Sfriso, A.; Pugnetti, A. Studies of the spatial and temporal variability of microphytobenthos in the Venice Lagoon. In Scientific Research and Safeguarding of Venice (Corila Research Program 2004–2006, 2005 Results); Vol, V., Campostrini, P., Eds.; IVSLA. Multigraf srl.: Spinea, Italy, 2007; pp. 255–260. [Google Scholar]
- Solidoro, C.; Bandelj, V.; Bernardi, F.A.; Camatti, E.; Ciavatta, S.; Cossarini, G.; Facca, C.; Franzoi, P.; Libralato, S.; Melaku Canu, D.; et al. Response of Venice lagoon ecosystem to natural and anthropogenic pressures over the last 50 years. In Coastal Lagoons: Critical Habitats and Environmental Change, 1st ed.; Kennish, M., Paerl, H., Eds.; CRC Press, Taylor and Francis: Boca Raton, FL, USA, 2010; pp. 483–511. [Google Scholar] [CrossRef]
- Acri, F.; Bernardi-Aubry, F.; Berton, A.; Bianchi, F.; Boldrin, A.; Camatti, E.; Comaschi, A.; Rabitti, S.; Socal, G. Plankton communities and nutrients in the Venice Lagoon: Comparison between current and old data. J. Mar. Syst. 2004, 51, 321–329. [Google Scholar] [CrossRef]
- Facca, C.; Sfriso, A.; Socal, G. Changes in abundance and composition of phytoplankton and microphytobenthos due to increased sediment fluxes in the Venice Lagoon, Italy. Estuar Coast Shelf Sci. 2002, 54, 773–792. [Google Scholar] [CrossRef]
- Facca, C.; Sfriso, A. Phytoplankton in a transitional ecosystem of the Northern Adriatic Sea and its putative role as an indicator for water quality assessment. Mar Ecol. 2009, 30, 462–479. [Google Scholar] [CrossRef]
- Sfriso, A.; Facca, C.; Ghetti, P.F. Validation of the Macrophyte Quality Index (MaQI) set up to assess the ecological status of Italian marine transitional environments. Hydrobiologia 2009, 617, 117–141. [Google Scholar] [CrossRef]
- Masiol, M.; Facca, C.; Visin, F.; Sfriso, A.; Pavoni, B. Inter-annual heavy element and nutrient concentration trends in the top sediments of Venice Lagoon (Italy). Mar. Pollut. Bull. 2014, 89, 49–58. [Google Scholar] [CrossRef]
- Raccanelli, S.; Libralato, S.; Favotto, M. On the detoxification of benthic bivalve contaminated by POPs: Insights from experimental and modelling approaches. Environ. Chem. Lett. 2008, 6, 251–258. [Google Scholar] [CrossRef]
- Guerzoni, S.; Raccanelli, S. Observations on the Dioxin and the Other Organic Persistent Pollutant (POP) in Venice. In The Wounded Lagoon, 1st ed.; Cacciari, P., Da Villa, E., Eds.; Libreria Editrice Cafoscarina: Venezia, Italy, 2003; pp. 17–24. (In Italian) [Google Scholar]
- Sfriso, A.; Facca, C.; Sonia, C.; Marcomini, A. Recording the occurrence of trophic level changes in the lagoon of Venice over the 90s. Environ. Int. 2005, 31, 993–1001. [Google Scholar] [CrossRef]
- Sfriso, A.; Argese, E.; Bettiol, C.; Facca, C. Tapes philippinarum seed exposure to metals in polluted areas of the Venice lagoon. Estuar. Coast. Mar. Sci. 2008, 79, 581–590. [Google Scholar] [CrossRef]
- Weiss, R. The solubility of nitrogen, oxygen and argon in water and seawater. Deep-Sea Res. Oceanogr. Abstr. 1970, 17, 721–735. [Google Scholar] [CrossRef]
- Strickland, J.D.H.; Parson, T.R. A Practical Handbook of Seawater Analyses, 2nd ed.; Fisheries Research Board: Ottawa, ON, Canada, 1972. [Google Scholar]
- Lorenzen, C.J. Determination of chlorophyll and pheopigments: Spectrophotometric equations. Limnol. Oceanogr. 1967, 12, 343–346. [Google Scholar] [CrossRef]
- Oxner, M.R. The Determination of Chlorinity by the Knudsen Method and Hydrographical Tables; G.M. Manufacturing Co.: New York, NY, USA, 1962; p. 63. [Google Scholar]
- Sfriso, A.; Facca, C.; Bonometto, A.; Boscolo Brusà, R. Compliance of the macrophyte quality index (MaQI) with the WFD (2000/60/EC) and ecological status assessment in transitional areas: The Venice lagoon as study case. Ecol. Indic. 2014, 46, 536–547. [Google Scholar] [CrossRef]
- Mejia, A.Y.; Rotini, A.; Lacasella, F.; Bookman, R.; Thaller, M.C.; Shem-Tov, R.; Winters, G.; Migliore, L. Assessing the ecological status of seagrasses using morphology, biochemical descriptors and microbial community analyses. A study in Halophila stipulacea (Forsk.) Aschers meadows in the northern Red Sea. Ecol. Indic. 2016, 60, 1150–1163. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acid Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand J. Stat. 1984, 11, 265–270. [Google Scholar]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst—a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acid Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Clarke, K.R.; Warwick, R.M. Changes in Marine Communities: An Approach to Statistical Analyses and Interpretation; PRIMER-E: Plymouth, UK, 1994. [Google Scholar]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 25 June 2020).
- Clarke, K.R.; Gorley, R.N. PRIMER v6: User Manual/Tutorial (Plymouth Routines in Multivariate Ecological Research); PRIMER-E: Plymouth, UK, 2006. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protocols. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Serebryakova, A.; Aires, T.; Viard, F.; Serrão, E.A.; Engelen, A.H. Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PLoS ONE 2018, 13, e0206734. [Google Scholar] [CrossRef]
- Burke, C.; Thomas, T.; Lewis, M.; Steinberg, P.; Kjelleberg, S. Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. Isme J. 2011, 5, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Florez, J.Z.; Camus, C.; Hengst, M.B.; Buschmann, A.H. A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front. Microbiol. 2017, 8, 2561. [Google Scholar] [CrossRef]
- Elifantz, H.; Horn, G.; Ayon, M.; Cohen, Y.; Minz, D. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean coastal seawater. Fems Microbiol. Ecol. 2013, 85, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Buchan, A.; González, J.M.; Moran, M.A. Overview of the marine roseobacter lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.; Webb, J.S.; Holmström, C.; Case, R.; Low, A.; Steinberg, P.; Kjelleberg, S. Low densities epiphytic bacteria from the marine alga Ulva australis inhibit settlement of fouling organisms. Appl. Environ. Microbiol. 2007, 73, 7844–7852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Lu, G.; Zheng, Y.; Xie, W.; Li, S.; Hu, Z. Aquimarina agarilytica sp. nov., an agarolytic species isolated from a red alga. Int. J. Syst. Evol. Microbiol. 2012, 62, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigel, B.L.; Pfister, C.A. Successional dynamics and seascape-level patterns of microbial communities on the canopy-forming kelps Nereocystis luetkeana and Macrocystis pyrifera. Front. Microbiol. 2019, 10, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, L.N.; Hutchison, K.; Grossman, A.R.; Brawley, S.H. Diversity and abundance of the bacterial community of the red macroalga Porphyra umbilicalis: Did bacterial farmers produce macroalgae? PLoS ONE 2013, 8, e58269. [Google Scholar] [CrossRef]
- McIlroy, S.J.; Nielsen, P.H. The Family Saprospiraceae. In The Prokaryotes, 1st ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 863–889. [Google Scholar] [CrossRef]
- Mancuso, F.P.; D’Hondt, S.; Willems, A.; Airoldi, L.; De Clerck, O. Diversity and temporal dynamics of the epiphytic bacterial communities associated with the canopy-forming seaweed Cystoseira compressa (Esper) Gerloff and Nizamuddin. Front. Microbiol. 2016, 7, 476. [Google Scholar] [CrossRef] [Green Version]
- Aires, T.; Muyzer, G.; Serrão, E.A.; Engelen, A.H. Seaweed Loads Cause Stronger Bacterial Community Shifts in Coastal Lagoon Sediments Than Nutrient Loads. Front. Microbiol. 2019, 9, 3283. [Google Scholar] [CrossRef]
- Rivett, D.W.; Jones, M.L.; Ramoneda, J.; Mombrikotb, S.B.; Ransome, E.; Bell, T. Elevated success of multispecies bacterial invasions impacts community composition during ecological succession. Ecol. Lett. 2018, 21, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, K.; Schindler, D.E.; Horner-Devine, M.C. Resource availability and spatial heterogeneity control bacterial community response to nutrient enrichment in lakes. PLoS ONE 2014, 9, e86991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raes, E.J.; Bodrossy, L.; van de Kamp, J.; Bissett, A.; Waite, A.M. Marine bacterial richness increases towards higher latitudes in the eastern Indian Ocean. Limnol. Oceanogr. Lett. 2018, 3, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Losso, C.; Ghirardini, A.V. Overview of ecotoxicological studies performed in the Venice Lagoon (Italy). Environ. Int. 2010, 36, 92–121. [Google Scholar] [CrossRef] [PubMed]
- Kolmonen, E.; Sivonen, K.; Rapala, J.; Haukka, K. Diversity of cyanobacteria and heterotrophic bacteria in cyanobacterial blooms in Lake Joutikas, Finland. Aquat. Microb. Ecol. 2004, 36, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Zozaya-Valdes, E.; Egan, S.; Thomas, T. A comprehensive analysis of the microbial communities of healthy and diseased marine macroalgae and the detection of known and potential bacterial pathogens. Front. Microbiol. 2015, 6, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedashkovskaya, O.I.; Kukhlevskiy, A.D.; Zhukova, N.V.; Kim, S.J.; Rhee, S.K. Litorimonas cladophorae sp. nov., a new alphaproteobacterium isolated from the Pacific green alga Cladophora stimpsoni, and emended descriptions of the genus Litorimonas and Litorimonas taeaensis. Antonie Van Leeuwenhoek 2013, 103, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Fulton, R.S., 3rd; Moisander, P.H.; Dyble, J. Harmful freshwater algal blooms, with an emphasis on cyanobacteria. Sci. World J. 2001, 1, 76–113. [Google Scholar] [CrossRef]
- Millie, D.F.; Weckman, G.R.; Fahnenstiel, G.L.; Carrick, H.J.; Ardjmand, E.; Young, W.A.; Sayers, M.J.; Shuchman, R.A. Using artificial intelligence for CyanoHAB niche modeling: Discovery and visualization of Microcystis—Environmental associations within western Lake Erie. Can. J. Fish. Aquat. Sci. 2014, 71, 1642–1654. [Google Scholar] [CrossRef] [Green Version]
- Lage, O.M.; Bondoso, J. Planctomycetes and macroalgae, a striking association. Front. Microbiol. 2014, 5, 267. [Google Scholar] [CrossRef] [Green Version]
- Jurelevicius, D.; Alvarez, V.M.; Marques, J.M.; de Sousa Lima, L.R.; Dias, F.; Seldin, L. Bacterial community response to petroleum hydrocarbon amendments in freshwater, marine, and hypersaline water-containing microcosms. Appl. Environ. Microbiol. 2013, 79, 5927–5935. [Google Scholar] [CrossRef] [Green Version]
- Kostka, J.E.; Prakash, O.; Overholt, W.A.; Green, S.J.; Freyer, G.; Canion, A.; Delgardio, J.; Norton, N.; Hazen, T.C.; Huettel, M. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl. Environ. Microbiol. 2011, 77, 7962–7974. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Steele, J.A.; Coapraso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Susan Huse, S.; McHardy, A.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. Isme J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luria, C.M.; Amaral-Zettler, L.A.; Ducklow, H.W.; Rich, J.J. Seasonal succession of free-living bacterial communities in coastal waters of the western Antarctic peninsula. Front. Microbiol. 2016, 7, 1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemura, A.; Chien, D.; Polz, M. Associations and dynamics of Vibrionaceae in the environment, from the genus to the population level. Front. Microbiol. 2014, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Minich, J.J.; Morris, M.M.; Brown, M.; Doane, M.; Edwards, M.S.; Michael, T.P.; Dinsdale, E.A. Elevated temperature drives kelp microbiome dysbiosis, while elevated carbon dioxide induces water microbiome disruption. PLoS ONE 2018, 13, e0192772. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Liu, B.; Braker, G. Isolation, genetic and functional characterization of novel soil nirK-type denitrifiers. Syst. Appl. Microbiol. 2010, 33, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Eloe, E.A.; Shulse, C.N.; Fadrosh, D.W.; Williamson, S.J.; Allen, E.E.; Bartlett, D.H. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 2011, 3, 449–458. [Google Scholar] [CrossRef]
- Aires, T.; Serebryakova, A.; Viard, F.; Serrão, E.A.; Engelen, A.H. Acidification increases abundances of Vibrionales and Planctomycetia associated to a seaweed-grazer system: Potential consequences for disease and prey digestion efficiency. Peer J. 2018, 6, e4377. [Google Scholar] [CrossRef] [Green Version]
- Witt, V.; Wild, C.; Anthony, K.R.N.; Diaz-Pulido, G.; Uthicke, S. Effects of ocean acidification on microbial community composition of, and oxygen fluxes through, biofilms from the Great Barrier Reef. Environ. Microbiol. 2011, 13, 2976–2989. [Google Scholar] [CrossRef]
- Verbaendert, I.; Boon, N.; De Vos, P.; Heylen, K. Denitrification is a common feature among members of the genus Bacillus. Syst. Appl. Microbiol. 2011, 34, 385–391. [Google Scholar] [CrossRef]
- Bellucci, L.G.; Frignani, M.; Paolucci, D.; Ravanelli, M. Distribution of heavy metals in sediments of the Venice Lagoon: The role of the industrial area. Sci. Total Environ. 2002, 295, 35–49. [Google Scholar] [CrossRef]
- Zonta, R.; Botter, M.; Cassin, D.; Pini, R.; Scattolin, M.; Zaggia, L. Sediment chemical contamination of a shallow water area close to the industrial zone of Porto Marghera (Venice Lagoon, Italy). Mar. Pollut. Bull. 2007, 55, 529–542. [Google Scholar] [CrossRef]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Baussant, T. Capturing Early Changes in the Marine Bacterial Community as a Result of Crude Oil Pollution in a Mesocosm Experiment. Microbes Environ. 2017, 32, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Ma, Y.; Wei, C.; Jiao, N.; Tang, K.; Wu, Z.; Jian, J. Bacterial diversity in various coastal mariculture ponds in Southeast China and in diseased eels as revealed by culture and culture-independent molecular techniques. Aquac. Res. 2010, 41, e172–e186. [Google Scholar] [CrossRef]
- Vincent, P.; Pignet, P.; Talmont, F.; Bozzi, L.; Fournet, B.; Guezennec, J.; Prieur, D. Production and characterization of an exo-polysaccharide excreted by a deep-sea hydrothermal vent bacterium isolated from the polychaete annelid Alvinella pompejana. Appl. Environ. Microbiol. 1994, 60, 4134–4141. [Google Scholar] [CrossRef] [Green Version]
- Neave, M.J.; Streten-Joyce, C.; Glasby, C.J.; McGuinness, K.A.; Parry, D.L.; Gibb, K.S. The bacterial community associated with the marine Polychaete ophelina sp. (Annelida: Opheliidae) is altered by copper and zinc contamination in sediments. Microb. Ecol. 2012, 63, 639–650. [Google Scholar] [CrossRef]
- Wahsha, M.; Juhmani, A.F.; Buosi, A.; Sfriso, A.; Sfriso, A. Assess the environmental health status of macrophyte ecosystems using an oxidative stress biomarker. Case studies: The Gulf of Aqaba and the Lagoon of Venice. Energy Procedia 2017, 125, 19–26. [Google Scholar] [CrossRef]
- Lamendella, R.; Strutt, S.; Borglin, S.; Chakraborty, R.; Tas, N.; Mason, O.; Hultman, J.; Prestat, E.; Hazen, T.; Jansson, J. Assessment of the Deepwater Horizon oil spill impact on Gulf coast microbial communities. Front. Microbiol. 2014, 5, 130. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Season | pH | Eh (mv) | DO (%) | Sal. (PSU) | Temp. (°C) | Chl-a (µg/L) |
|---|---|---|---|---|---|---|---|
| SMM | Winter | 8.37 | 298 | 109.9 | 24.9 | 14.2 | 2.08 |
| Spring | 8.40 | 259 | 111.1 | 22.2 | 20.5 | 0.75 | |
| Summer | 8.32 | 265 | 110.4 | 32.5 | 25.2 | 0.60 | |
| Autumn | 8.10 | 312 | 99.3 | 27.0 | 19.0 | 0.75 | |
| PM | Winter | 8.15 | 307 | 111.2 | 25.4 | 15.8 | 1.39 |
| Spring | 8.33 | 219 | 118.0 | 23.7 | 25.5 | 1.50 | |
| Summer | 8.10 | 300 | 76.1 | 27.2 | 30.6 | 3.29 | |
| Autumn | 8.01 | 300 | 79.1 | 22.5 | 20.0 | 1.50 | |
| SG | Winter | 8.12 | 346 | 94.7 | 6.4 | 14.0 | 2.08 |
| Spring | 8.73 | 222 | 180.4 | 17.5 | 26.1 | 29.9 | |
| Summer | 8.11 | 570 | 87.0 | 26.5 | 29.8 | 3.59 | |
| Autumn | 7.91 | 297 | 76.8 | 26.6 | 19.3 | 1.80 |
| ASV | SMM | PM | SG | Phylum | Class | Order | Family | Genus |
|---|---|---|---|---|---|---|---|---|
| ASV1 | 0 | 6.70 | 0.46 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Nitrincolaceae | NA |
| ASV7 | 5.35 | 0.54 | <0.10 | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | NA |
| ASV9 | 0 | 3.06 | 0.53 | Proteobacteria | Alphaproteobacteria | Caulobacterales | Hyphomonadaceae | Litorimonas |
| ASV17 | 1.62 | 1.18 | 0.68 | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | Polaribacter |
| ASV12 | <0.10 | 3.20 | 0.27 | Proteobacteria | Alphaproteobacteria | Caulobacterales | Hyphomonadaceae | Hellea |
| ASV5 | 1.47 | 1.32 | 0.64 | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | NA |
| ASV21 | 2.84 | 0 | <0.10 | Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Rubritaleaceae | NA |
| ASV10 | 0 | 2.52 | 0.16 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Nitrincolaceae | NA |
| ASV13 | 0 | 2.36 | 0.15 | Proteobacteria | Gammaproteobacteria | Oceanospirillales | Nitrincolaceae | NA |
| ASV27 | 2.20 | 0.23 | <0.10 | Bacteroidetes | Bacteroidia | Flavobacteriales | Flavobacteriaceae | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juhmani, A.-S.; Vezzi, A.; Wahsha, M.; Buosi, A.; Pascale, F.D.; Schiavon, R.; Sfriso, A. Diversity and Dynamics of Seaweed Associated Microbial Communities Inhabiting the Lagoon of Venice. Microorganisms 2020, 8, 1657. https://doi.org/10.3390/microorganisms8111657
Juhmani A-S, Vezzi A, Wahsha M, Buosi A, Pascale FD, Schiavon R, Sfriso A. Diversity and Dynamics of Seaweed Associated Microbial Communities Inhabiting the Lagoon of Venice. Microorganisms. 2020; 8(11):1657. https://doi.org/10.3390/microorganisms8111657
Chicago/Turabian StyleJuhmani, Abdul-Salam, Alessandro Vezzi, Mohammad Wahsha, Alessandro Buosi, Fabio De Pascale, Riccardo Schiavon, and Adriano Sfriso. 2020. "Diversity and Dynamics of Seaweed Associated Microbial Communities Inhabiting the Lagoon of Venice" Microorganisms 8, no. 11: 1657. https://doi.org/10.3390/microorganisms8111657
APA StyleJuhmani, A. -S., Vezzi, A., Wahsha, M., Buosi, A., Pascale, F. D., Schiavon, R., & Sfriso, A. (2020). Diversity and Dynamics of Seaweed Associated Microbial Communities Inhabiting the Lagoon of Venice. Microorganisms, 8(11), 1657. https://doi.org/10.3390/microorganisms8111657