The Role of APOBECs in Viral Replication


Abstract
:1. Introduction
1.1. APOBEC Enzyme Activity
1.2. APOBEC Protein Structures
1.2.1. Nucleotide-Binding Activity
1.2.2. APOBEC Domain Structure
1.3. APOBEC Expression and Localization
2. Viral Restriction by APOBEC Enzymes
2.1. Deaminase-Dependent Inhibition of Viruses by APOBECs
2.1.1. HIV-1 Hypermutation by A3 Enzymes in Cell Culture
2.1.2. A3-Mediated Hypermutation in HIV-1-Infected Patients
2.1.3. Vif Antagonism of A3 Proteins
2.1.4. Deamination-Dependent Hypermutation of HTLV-1
2.1.5. Deamination-Dependent A3 Activity and Murine Retroviruses
2.1.6. Deamination of Other RNA-Containing Viruses by APOBECs
2.1.7. Deamination-Dependent Antagonism of DNA-Containing Viruses
2.2. Deamination-Independent Antagonism of Viruses by APOBEC Enzymes
2.2.1. Deamination-Independent Inhibition of HIV-1
2.2.2. Betaretroviruses and Deamination-Independent APOBEC Activity
2.2.3. Deamination-Independent Inhibition of Murine Leukemia Viruses
2.2.4. Deamination-Independent Inhibition of Other RNA-Containing Viruses
2.2.5. Deaminase-Independent Antagonism of DNA-Containing Viruses
3. APOBEC Proteins and Restriction of Endogenous Viruses and Retrotransposons
3.1. ERVs and APOBEC-Mediated Inhibition
3.2. Non-LTR Retrotransposons and APOBECs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.E.; Harris, R.S.; Harki, D.A. APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem. Biol. 2018, 25, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conticello, S.G.; Thomas, C.J.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzysiak, T.C.; Jung, J.; Thompson, J.; Baker, D.; Gronenborn, A.M. APOBEC2 is a monomer in solution: Implications for APOBEC3G models. Biochemistry 2012, 51, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.P.; Lin, J.J.; Hu, C.Y.; Hsu, Y.H.; Wang, A.H.; Liaw, S.H. Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J. Biol. Chem. 2003, 278, 19111–19117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireton, G.C.; Black, M.E.; Stoddard, B.L. The 1.14 A crystal structure of yeast cytosine deaminase: Evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure 2003, 11, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Sowden, M.P.; Dance, G.S.; Torelli, A.T.; Smith, H.C.; Wedekind, J.E. The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc. Natl. Acad. Sci. USA 2004, 101, 8114–8119. [Google Scholar] [CrossRef] [Green Version]
- Betts, L.; Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W., Jr. Cytidine deaminase. The 2.3 A crystal structure of an enzyme: Transition-state analog complex. J. Mol. Biol. 1994, 235, 635–656. [Google Scholar] [CrossRef]
- Johansson, E.; Mejlhede, N.; Neuhard, J.; Larsen, S. Crystal structure of the tetrameric cytidine deaminase from Bacillus subtilis at 2.0 A resolution. Biochemistry 2002, 41, 2563–2570. [Google Scholar] [CrossRef]
- Shandilya, S.M.; Nalam, M.N.; Nalivaika, E.A.; Gross, P.J.; Valesano, J.C.; Shindo, K.; Li, M.; Munson, M.; Royer, W.E.; Harjes, E.; et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 2010, 18, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Bostrom, K.; Garcia, Z.; Poksay, K.S.; Johnson, D.F.; Lusis, A.J.; Innerarity, T.L. Apolipoprotein B mRNA editing. Direct determination of the edited base and occurrence in non-apolipoprotein B-producing cell lines. J. Biol. Chem. 1990, 265, 22446–22452. [Google Scholar] [PubMed]
- Lerner, T.; Papavasiliou, F.N.; Pecori, R. RNA Editors, Cofactors, and mRNA Targets: An Overview of the C-to-U RNA Editing Machinery and Its Implication in Human Disease. Genes 2018, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, J.D.; Smith, H.C. Modeling the Embrace of a Mutator: APOBEC Selection of Nucleic Acid Ligands. Trends Biochem. Sci. 2018, 43, 606–622. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, B.R.; Hamilton, C.E.; Mwangi, M.M.; Dewell, S.; Papavasiliou, F.N. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat. Struct. Mol. Biol. 2011, 18, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Lellek, H.; Kirsten, R.; Diehl, I.; Apostel, F.; Buck, F.; Greeve, J. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J. Biol. Chem. 2000, 275, 19848–19856. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Kinter, M.T.; Sherman, N.E.; Driscoll, D.M. Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol. Cell. Biol. 2000, 20, 1846–1854. [Google Scholar] [CrossRef] [Green Version]
- Fossat, N.; Tourle, K.; Radziewic, T.; Barratt, K.; Liebhold, D.; Studdert, J.B.; Power, M.; Jones, V.; Loebel, D.A.; Tam, P.P. C to U RNA editing mediated by APOBEC1 requires RNA-binding protein RBM47. EMBO Rep. 2014, 15, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R.; Knott, T.J.; Legros, J.E.; Navaratnam, N.; Greeve, J.C.; Scott, J. Sequence requirements for the editing of apolipoprotein B mRNA. J. Biol. Chem. 1991, 266, 16301–16304. [Google Scholar]
- Backus, J.W.; Smith, H.C. Apolipoprotein B mRNA sequences 3′ of the editing site are necessary and sufficient for editing and editosome assembly. Nucleic Acids Res. 1991, 19, 6781–6786. [Google Scholar] [CrossRef] [Green Version]
- Beale, R.C.; Petersen-Mahrt, S.K.; Watt, I.N.; Harris, R.S.; Rada, C.; Neuberger, M.S. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: Correlation with mutation spectra in vivo. J. Mol. Biol. 2004, 337, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Petersen-Mahrt, S.K.; Neuberger, M.S. In vitro deamination of cytosine to uracil in single-stranded DNA by apolipoprotein B editing complex catalytic subunit 1 (APOBEC1). J. Biol. Chem. 2003, 278, 19583–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjes, S.; Solomon, W.C.; Li, M.; Chen, K.M.; Harjes, E.; Harris, R.S.; Matsuo, H. Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 2013, 87, 7008–7014. [Google Scholar] [CrossRef] [Green Version]
- Nabel, C.S.; Lee, J.W.; Wang, L.C.; Kohli, R.M. Nucleic acid determinants for selective deamination of DNA over RNA by activation-induced deaminase. Proc. Natl. Acad. Sci. USA 2013, 110, 14225–14230. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, M.A.; Rajagurubandara, E.; Wijesinghe, P.; Bhagwat, A.S. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair 2010, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Maul, R.W.; Guminski, A.F.; McClure, R.L.; Gajula, K.S.; Saribasak, H.; McMahon, M.A.; Siliciano, R.F.; Gearhart, P.J.; Stivers, J.T. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J. Biol. Chem. 2010, 285, 40956–40964. [Google Scholar] [CrossRef] [Green Version]
- Rathore, A.; Carpenter, M.A.; Demir, O.; Ikeda, T.; Li, M.; Shaban, N.M.; Law, E.K.; Anokhin, D.; Brown, W.L.; Amaro, R.E.; et al. The local dinucleotide preference of APOBEC3G can be altered from 5′ -CC to 5′ -TC by a single amino acid substitution. J. Mol. Biol. 2013, 425, 4442–4454. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Rada, C.; Neuberger, M.S. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J. Exp. Med. 2010, 207, 141–153. [Google Scholar] [CrossRef]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Kurt Yilmaz, N.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, O.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Chelico, L.; Goodman, M.F.; Le Grice, S.F. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J. Biol. Chem. 2009, 284, 7047–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtz, C.M.; Sadler, H.A.; Mansky, L.M. APOBEC3G cytosine deamination hotspots are defined by both sequence context and single-stranded DNA secondary structure. Nucleic Acids Res. 2013, 41, 6139–6148. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Q.; Wang, L.; Meng, F.L.; Hwang, J.K.; Alt, F.W.; Wu, H. AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 2017, 67, 361–373.e364. [Google Scholar] [CrossRef]
- Salter, J.D.; Polevoda, B.; Bennett, R.P.; Smith, H.C. Regulation of Antiviral Innate Immunity Through APOBEC Ribonucleoprotein Complexes. Subcell. Biochem. 2019, 93, 193–219. [Google Scholar] [CrossRef]
- Fang, Y.; Xiao, X.; Li, S.X.; Wolfe, A.; Chen, X.S. Molecular Interactions of a DNA Modifying Enzyme APOBEC3F Catalytic Domain with a Single-Stranded DNA. J. Mol. Biol. 2018, 430, 87–101. [Google Scholar] [CrossRef]
- McDougall, W.M.; Smith, H.C. Direct evidence that RNA inhibits APOBEC3G ssDNA cytidine deaminase activity. Biochem. Biophys. Res. Commun. 2011, 412, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Polevoda, B.; McDougall, W.M.; Tun, B.N.; Cheung, M.; Salter, J.D.; Friedman, A.E.; Smith, H.C. RNA binding to APOBEC3G induces the disassembly of functional deaminase complexes by displacing single-stranded DNA substrates. Nucleic Acids Res. 2015, 43, 9434–9445. [Google Scholar] [CrossRef] [Green Version]
- McDougall, W.M.; Okany, C.; Smith, H.C. Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J. Biol. Chem. 2011, 286, 30655–30661. [Google Scholar] [CrossRef] [Green Version]
- Opi, S.; Takeuchi, H.; Kao, S.; Khan, M.A.; Miyagi, E.; Goila-Gaur, R.; Iwatani, Y.; Levin, J.G.; Strebel, K. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 2006, 80, 4673–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog 2006, 2, e41. [Google Scholar] [CrossRef] [PubMed]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef] [Green Version]
- Shaban, N.M.; Shi, K.; Lauer, K.V.; Carpenter, M.A.; Richards, C.M.; Salamango, D.; Wang, J.; Lopresti, M.W.; Banerjee, S.; Levin-Klein, R.; et al. The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol. Cell 2018, 69, 75–86.e79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, F.; Yang, H.; Xiao, X.; Li, S.X.; Wolfe, A.; Zirkle, B.; Arutiunian, V.; Chen, X.S. Understanding the Structure, Multimerization, Subcellular Localization and mC Selectivity of a Genomic Mutator and Anti-HIV Factor APOBEC3H. Sci. Rep. 2018, 8, 3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Wong, L.; Morse, M.; Rouzina, I.; Williams, M.C.; Chelico, L. RNA-Mediated Dimerization of the Human Deoxycytidine Deaminase APOBEC3H Influences Enzyme Activity and Interaction with Nucleic Acids. J. Mol. Biol. 2018, 430, 4891–4907. [Google Scholar] [CrossRef]
- Bohn, J.A.; Thummar, K.; York, A.; Raymond, A.; Brown, W.C.; Bieniasz, P.D.; Hatziioannou, T.; Smith, J.L. APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat. Commun. 2017, 8, 1021. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Tian, C.; Zhang, W.; Sarkis, P.T.; Yu, X.F. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J. Mol. Biol. 2008, 375, 1098–1112. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, W.; Tian, C.; Liu, B.; Yu, Y.; Ding, L.; Spearman, P.; Yu, X.F. Distinct viral determinants for the packaging of human cytidine deaminases APOBEC3G and APOBEC3C. Virology 2008, 377, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.; Peddi, S.; Mangeat, B.; Lakkaraju, A.; Strub, K.; Trono, D. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 2008, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- York, A.; Kutluay, S.B.; Errando, M.; Bieniasz, P.D. The RNA Binding Specificity of Human APOBEC3 Proteins Resembles That of HIV-1 Nucleocapsid. PLoS Pathog. 2016, 12, e1005833. [Google Scholar] [CrossRef] [PubMed]
- Silvas, T.V.; Schiffer, C.A. APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci. 2019, 28, 1552–1566. [Google Scholar] [CrossRef] [PubMed]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.C. RNA binding to APOBEC deaminases; Not simply a substrate for C to U editing. RNA Biol. 2017, 14, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012, 12, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Abudu, A.; Son, S.; Dang, Y.; Venta, P.J.; Zheng, Y.H. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 2011, 85, 3142–3152. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Carpenter, M.A.; Kurahashi, K.; Harris, R.S.; Aihara, H. Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J. Biol. Chem. 2015, 290, 28120–28130. [Google Scholar] [CrossRef] [Green Version]
- Marx, A.; Galilee, M.; Alian, A. Zinc enhancement of cytidine deaminase activity highlights a potential allosteric role of loop-3 in regulating APOBEC3 enzymes. Sci. Rep. 2015, 5, 18191. [Google Scholar] [CrossRef]
- Bohn, M.F.; Shandilya, S.M.D.; Silvas, T.V.; Nalivaika, E.A.; Kouno, T.; Kelch, B.A.; Ryder, S.P.; Kurt-Yilmaz, N.; Somasundaran, M.; Schiffer, C.A. The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 2015, 23, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Myint, W.; Kanai, T.; Delviks-Frankenberry, K.; Sierra Rodriguez, C.; Pathak, V.K.; Schiffer, C.A.; Matsuo, H. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018, 9, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, F.; Bollman, B.; Chen, H.; Konig, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Zhang, T.; Xu, Z.; Liu, S.; Zhao, B.; Lan, W.; Wang, C.; Ding, J.; Cao, C. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 2015, 290, 4010–4021. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Li, S.X.; Yang, H.; Chen, X.S. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016, 7, 12193. [Google Scholar] [CrossRef]
- Yang, H.; Ito, F.; Wolfe, A.D.; Li, S.; Mohammadzadeh, N.; Love, R.P.; Yan, M.; Zirkle, B.; Gaba, A.; Chelico, L.; et al. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat. Commun. 2020, 11, 632. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, S.; Marusawa, H.; Matsumoto, T.; Ueda, Y.; Matsumoto, Y.; Endo, Y.; Takai, A.; Chiba, T. Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis. Int. J. Cancer 2012, 130, 1294–1301. [Google Scholar] [CrossRef] [Green Version]
- Prochnow, C.; Bransteitter, R.; Klein, M.G.; Goodman, M.F.; Chen, X.S. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 2007, 445, 447–451. [Google Scholar] [CrossRef]
- Lada, A.G.; Krick, C.F.; Kozmin, S.G.; Mayorov, V.I.; Karpova, T.S.; Rogozin, I.B.; Pavlov, Y.I. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry 2011, 76, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, Y.; Li, M.; Carpenter, M.A.; McDougle, R.M.; Luengas, E.M.; Macdonald, P.J.; Harris, R.S.; Mueller, J.D. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J. Mol. Biol. 2014, 426, 1296–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, B.; Burant, C.F.; Davidson, N.O. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 1993, 260, 1816–1819. [Google Scholar] [CrossRef] [PubMed]
- Harjanto, D.; Papamarkou, T.; Oates, C.J.; Rayon-Estrada, V.; Papavasiliou, F.N.; Papavasiliou, A. RNA editing generates cellular subsets with diverse sequence within populations. Nat. Commun. 2016, 7, 12145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Oynebraten, I.; Corthay, A. Toll-Like Receptor Ligands and Interferon-gamma Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef]
- Cole, D.C.; Chung, Y.; Gagnidze, K.; Hajdarovic, K.H.; Rayon-Estrada, V.; Harjanto, D.; Bigio, B.; Gal-Toth, J.; Milner, T.A.; McEwen, B.S.; et al. Loss of APOBEC1 RNA-editing function in microglia exacerbates age-related CNS pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 13272–13277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Ohtsubo, H.; Nihei, N.; Kaneko, T.; Sato, Y.; Adachi, S.I.; Kondo, S.; Nakamura, M.; Mizunoya, W.; Iida, H.; et al. Apobec2 deficiency causes mitochondrial defects and mitophagy in skeletal muscle. FASEB J. 2018, 32, 1428–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Probst, H.C.; Tatsumi, R.; Ikeuchi, Y.; Neuberger, M.S.; Rada, C. Deficiency in APOBEC2 leads to a shift in muscle fiber type, diminished body mass, and myopathy. J. Biol. Chem. 2010, 285, 7111–7118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Mohanram, V.; Skold, A.E.; Bachle, S.M.; Pathak, S.K.; Spetz, A.L. IFN-alpha induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J. Immunol. 2013, 190, 3346–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisblum, Y.; Oiknine-Djian, E.; Zakay-Rones, Z.; Vorontsov, O.; Haimov-Kochman, R.; Nevo, Y.; Stockheim, D.; Yagel, S.; Panet, A.; Wolf, D.G. APOBEC3A Is Upregulated by Human Cytomegalovirus (HCMV) in the Maternal-Fetal Interface, Acting as an Innate Anti-HCMV Effector. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Lei, K.J.; Jin, W.; Greenwell-Wild, T.; Wahl, S.M. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 2006, 203, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Tau, G.; Rothman, P. Biologic functions of the IFN-gamma receptors. Allergy 1999, 54, 1233–1251. [Google Scholar] [CrossRef]
- Graziano, F.; Vicenzi, E.; Poli, G. Plastic restriction of HIV-1 replication in human macrophages derived from M1/M2 polarized monocytes. J. Leukoc. Biol. 2016, 100, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Cobos Jimenez, V.; Booiman, T.; de Taeye, S.W.; van Dort, K.A.; Rits, M.A.; Hamann, J.; Kootstra, N.A. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2012, 2, 763. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kajaste-Rudnitski, A.; Coradin, T.; Saba, E.; Della Chiara, G.; Barbagallo, M.; Graziano, F.; Alfano, M.; Cassol, E.; Vicenzi, E.; et al. M1 polarization of human monocyte-derived macrophages restricts pre and postintegration steps of HIV-1 replication. AIDS 2013, 27, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Covino, D.A.; Gauzzi, M.C.; Fantuzzi, L. Understanding the regulation of APOBEC3 expression: Current evidence and much to learn. J. Leukoc Biol. 2018, 103, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Pautasso, S.; Galitska, G.; Dell’Oste, V.; Biolatti, M.; Cagliani, R.; Forni, D.; De Andrea, M.; Gariglio, M.; Sironi, M.; Landolfo, S. Strategy of Human Cytomegalovirus To Escape Interferon Beta-Induced APOBEC3G Editing Activity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.P.; Salter, J.D.; Liu, X.; Wedekind, J.E.; Smith, H.C. APOBEC3G subunits self-associate via the C-terminal deaminase domain. J. Biol. Chem. 2008, 283, 33329–33336. [Google Scholar] [CrossRef] [Green Version]
- Zhen, A.; Du, J.; Zhou, X.; Xiong, Y.; Yu, X.F. Reduced APOBEC3H variant anti-viral activities are associated with altered RNA binding activities. PLoS ONE 2012, 7, e38771. [Google Scholar] [CrossRef]
- Abudu, A.; Takaori-Kondo, A.; Izumi, T.; Shirakawa, K.; Kobayashi, M.; Sasada, A.; Fukunaga, K.; Uchiyama, T. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol. 2006, 16, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hakata, Y.; Takeda, E.; Liu, Q.; Iwatani, Y.; Kozak, C.A.; Miyazawa, M. Two genetic determinants acquired late in Mus evolution regulate the inclusion of exon 5, which alters mouse APOBEC3 translation efficiency. PLoS Pathog. 2012, 8, e1002478. [Google Scholar] [CrossRef]
- Sanville, B.; Dolan, M.A.; Wollenberg, K.; Yan, Y.; Martin, C.; Yeung, M.L.; Strebel, K.; Buckler-White, A.; Kozak, C.A. Adaptive evolution of Mus Apobec3 includes retroviral insertion and positive selection at two clusters of residues flanking the substrate groove. PLoS Pathog. 2010, 6, e1000974. [Google Scholar] [CrossRef]
- Takeda, E.; Tsuji-Kawahara, S.; Sakamoto, M.; Langlois, M.A.; Neuberger, M.S.; Rada, C.; Miyazawa, M. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J. Virol. 2008, 82, 10998–11008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakata, Y.; Landau, N.R. Reversed functional organization of mouse and human APOBEC3 cytidine deaminase domains. J. Biol. Chem. 2006, 281, 36624–36631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeoma, C.M.; Huegel, A.L.; Lingappa, J.; Feldman, M.D.; Ross, S.R. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host Microbe 2010, 8, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Golovkina, T.V.; Dudley, J.P.; Ross, S.R. B and T cells are required for mouse mammary tumor virus spread within the mammary gland. J. Immunol. 1998, 161, 2375–2382. [Google Scholar] [PubMed]
- Bhattacharya, C.; Aggarwal, S.; Kumar, M.; Ali, A.; Matin, A. Mouse apolipoprotein B editing complex 3 (APOBEC3) is expressed in germ cells and interacts with dead-end (DND1). PLoS ONE 2008, 3, e2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, A. RNA Packaging in HIV. Trends Microbiol. 2019, 27, 715–723. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Basu, M.K.; Jordan, I.K.; Pavlov, Y.I.; Koonin, E.V. APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predicted by computational analysis. Cell Cycle 2005, 4, 1281–1285. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Tan, L.; Zhang, Y.; Meng, C.; Wang, W.; Sun, Y.; Song, C.; Liu, W.; Liao, Y.; Yu, S.; et al. Characterization and functional analysis of chicken APOBEC4. Dev. Comp. Immunol. 2020, 106, 103631. [Google Scholar] [CrossRef]
- Pone, E.J.; Zan, H.; Zhang, J.; Al-Qahtani, A.; Xu, Z.; Casali, P. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: Relevance to microbial antibody responses. Crit. Rev. Immunol. 2010, 30, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Cattoretti, G.; Buttner, M.; Shaknovich, R.; Kremmer, E.; Alobeid, B.; Niedobitek, G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood 2006, 107, 3967–3975. [Google Scholar] [CrossRef]
- Cantaert, T.; Schickel, J.N.; Bannock, J.M.; Ng, Y.S.; Massad, C.; Oe, T.; Wu, R.; Lavoie, A.; Walter, J.E.; Notarangelo, L.D.; et al. Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity 2015, 43, 884–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, J.A.; Castro, C.D.; Deiss, T.C.; Ohta, Y.; Flajnik, M.F.; Criscitiello, M.F. Somatic hypermutation of T cell receptor alpha chain contributes to selection in nurse shark thymus. elife 2018, 7, e28477. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Suzuki, K.; Nakata, M.; Chikuma, S.; Izumi, N.; Huong le, T.; Maruya, M.; Fagarasan, S.; Busslinger, M.; Honjo, T.; et al. Activation-induced cytidine deaminase expression in CD4+ T cells is associated with a unique IL-10-producing subset that increases with age. PLoS ONE 2011, 6, e29141. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Patenaude, A.M.; Affarel, B.; Lamarre, A.; Young, J.C.; Di Noia, J.M. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J. Exp. Med. 2010, 207, 2751–2765. [Google Scholar] [CrossRef] [Green Version]
- Orthwein, A.; Zahn, A.; Methot, S.P.; Godin, D.; Conticello, S.G.; Terada, K.; Di Noia, J.M. Optimal functional levels of activation-induced deaminase specifically require the Hsp40 DnaJa1. EMBO J. 2012, 31, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Hasler, J.; Rada, C.; Neuberger, M.S. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1alpha (eEF1A). Proc. Natl. Acad. Sci. USA 2011, 108, 18366–18371. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Liu, Y.; Wu, L.; Ma, X.; Liu, Q.; Huang, F.; Zhang, X.; Zhang, Y.; Zhang, J.; Luo, H.; et al. CUL7 E3 Ubiquitin Ligase Mediates the Degradation of Activation-Induced Cytidine Deaminase and Regulates the Ig Class Switch Recombination in B Lymphocytes. J. Immunol. 2019, 203, 269–281. [Google Scholar] [CrossRef]
- Patenaude, A.M.; Orthwein, A.; Hu, Y.; Campo, V.A.; Kavli, B.; Buschiazzo, A.; Di Noia, J.M. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat. Struct. Mol. Biol. 2009, 16, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, P.M.; Shaknovich, R. Epigenetic function of activation-induced cytidine deaminase and its link to lymphomagenesis. Front. Immunol. 2014, 5, 642. [Google Scholar] [CrossRef]
- Sodroski, J.; Goh, W.C.; Rosen, C.; Tartar, A.; Portetelle, D.; Burny, A.; Haseltine, W. Replicative and cytopathic potential of HTLV-III/LAV with sor gene deletions. Science 1986, 231, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Landau, N.R. Recent insights into HIV-1 Vif. Curr. Opin. Immunol. 2004, 16, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 2013, 87, 1508–1517. [Google Scholar] [CrossRef] [Green Version]
- Chaipan, C.; Smith, J.L.; Hu, W.S.; Pathak, V.K. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 2013, 87, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Chen, D.; Konig, R.; Mariani, R.; Unutmaz, D.; Landau, N.R. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 2004, 279, 53379–53386. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Brooks, A.; Chen, H.; Bennett, R.; Reichman, R.; Smith, H. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J. Virol. 2005, 79, 11513–11516. [Google Scholar] [CrossRef] [Green Version]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pasquale, M.; Kourteva, Y.; Allos, T.; D’Aquila, R.T. Lower HIV provirus levels are associated with more APOBEC3G protein in blood resting memory CD4+ T lymphocytes of controllers in vivo. PLoS ONE 2013, 8, e76002. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Iwabu, Y.; Tada, T.; Kawana-Tachikawa, A.; Koga, M.; Hosoya, N.; Nomura, S.; Brumme, Z.L.; Jessen, H.; Pereyra, F.; et al. Anti-APOBEC3G activity of HIV-1 Vif protein is attenuated in elite controllers. J. Virol. 2015, 89, 4992–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pery, E.; Sheehy, A.; Nebane, N.M.; Brazier, A.J.; Misra, V.; Rajendran, K.S.; Buhrlage, S.J.; Mankowski, M.K.; Rasmussen, L.; White, E.L.; et al. Identification of a novel HIV-1 inhibitor targeting Vif-dependent degradation of human APOBEC3G protein. J. Biol. Chem. 2015, 290, 10504–10517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, R.P.; Stewart, R.A.; Hogan, P.A.; Ptak, R.G.; Mankowski, M.K.; Hartman, T.L.; Buckheit, R.W., Jr.; Snyder, B.A.; Salter, J.D.; Morales, G.A.; et al. An analog of camptothecin inactive against Topoisomerase I is broadly neutralizing of HIV-1 through inhibition of Vif-dependent APOBEC3G degradation. Antivir. Res. 2016, 136, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Krisko, J.F.; Martinez-Torres, F.; Foster, J.L.; Garcia, J.V. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog. 2013, 9, e1003242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Izumi, T.; Misawa, N.; Kobayashi, T.; Yamashita, Y.; Ohmichi, M.; Ito, M.; Takaori-Kondo, A.; Koyanagi, Y. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J. Virol. 2010, 84, 9546–9556. [Google Scholar] [CrossRef] [Green Version]
- Luo, K.; Liu, B.; Xiao, Z.; Yu, Y.; Yu, X.; Gorelick, R.; Yu, X.F. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 2004, 78, 11841–11852. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.; Bogerd, H.P.; Cullen, B.R. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 2004, 328, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Svarovskaia, E.S.; Xu, H.; Mbisa, J.L.; Barr, R.; Gorelick, R.J.; Ono, A.; Freed, E.O.; Hu, W.S.; Pathak, V.K. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 2004, 279, 35822–35828. [Google Scholar] [CrossRef] [Green Version]
- Zennou, V.; Perez-Caballero, D.; Gottlinger, H.; Bieniasz, P.D. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Kao, S.; Miyagi, E.; Takeuchi, H.; Goila-Gaur, R.; Opi, S.; Gipson, C.L.; Parslow, T.G.; Ly, H.; Strebel, K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 2005, 79, 5870–5874. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Tian, C.; Zhang, W.; Luo, K.; Sarkis, P.T.; Yu, L.; Liu, B.; Yu, Y.; Yu, X.F. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 2007, 81, 13112–13124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.H.; Jeang, K.T.; Tokunaga, K. Host restriction factors in retroviral infection: Promises in virus-host interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Du, J.; Yu, K.; Wang, T.; Yong, X.; Yu, X.F. Association of potent human antiviral cytidine deaminases with 7SL RNA and viral RNP in HIV-1 virions. J. Virol. 2010, 84, 12903–12913. [Google Scholar] [CrossRef] [Green Version]
- Apolonia, L.; Schulz, R.; Curk, T.; Rocha, P.; Swanson, C.M.; Schaller, T.; Ule, J.; Malim, M.H. Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PLoS Pathog. 2015, 11, e1004609. [Google Scholar] [CrossRef] [Green Version]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 2014, 159, 1096–1109. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.; Spearman, P. APOBEC3G multimers are recruited to the plasma membrane for packaging into human immunodeficiency virus type 1 virus-like particles in an RNA-dependent process requiring the NC basic linker. J. Virol. 2007, 81, 5000–5013. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chertova, E.; Chen, J.; Ott, D.E.; Roser, J.D.; Hu, W.S.; Pathak, V.K. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 2007, 360, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Thangavelu, P.U.; Gupta, V.; Dixit, N.M. Estimating the fraction of progeny virions that must incorporate APOBEC3G for suppression of productive HIV-1 infection. Virology 2014, 449, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Chelico, L.; Sacho, E.J.; Erie, D.A.; Goodman, M.F. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008, 283, 13780–13791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Nagae, T.; Ode, H.; Awazu, H.; Kurosawa, T.; Hamano, A.; Matsuoka, K.; Hachiya, A.; Imahashi, M.; Yokomaku, Y.; et al. Structural basis of chimpanzee APOBEC3H dimerization stabilized by double-stranded RNA. Nucleic Acids Res. 2018, 46, 10368–10379. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrofelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suspene, R.; Rusniok, C.; Vartanian, J.P.; Wain-Hobson, S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006, 34, 4677–4684. [Google Scholar] [CrossRef]
- Wurtzer, S.; Goubard, A.; Mammano, F.; Saragosti, S.; Lecossier, D.; Hance, A.J.; Clavel, F. Functional central polypurine tract provides downstream protection of the human immunodeficiency virus type 1 genome from editing by APOBEC3G and APOBEC3B. J. Virol. 2006, 80, 3679–3683. [Google Scholar] [CrossRef] [Green Version]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef]
- Ikeda, T.; Molan, A.M.; Jarvis, M.C.; Carpenter, M.A.; Salamango, D.J.; Brown, W.L.; Harris, R.S. HIV-1 restriction by endogenous APOBEC3G in the myeloid cell line THP-1. J. Gen. Virol. 2019, 100, 1140–1152. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Burdick, R.C.; Izumi, T.; Doi, H.; Shao, W.; Alvord, W.G.; Sato, K.; Koyanagi, Y.; Jones, S.; Wilson, E.; et al. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res. 2016, 44, 7848–7865. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, D.; Anwar, F.; Davenport, M.P. APOBEC3G and APOBEC3F rarely co-mutate the same HIV genome. Retrovirology 2012, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krisko, J.F.; Begum, N.; Baker, C.E.; Foster, J.L.; Garcia, J.V. APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication. J. Virol. 2016, 90, 4681–4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [Green Version]
- Ara, A.; Love, R.P.; Follack, T.B.; Ahmed, K.A.; Adolph, M.B.; Chelico, L. Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Mohammadzadeh, N.; Follack, T.B.; Love, R.P.; Stewart, K.; Sanche, S.; Chelico, L. Polymorphisms of the cytidine deaminase APOBEC3F have different HIV-1 restriction efficiencies. Virology 2019, 527, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Prabhu, P.; Kenig, E.; Smith, Y.; Britan-Rosich, E.; Kotler, M. APOBEC3G inhibits HIV-1 RNA elongation by inactivating the viral trans-activation response element. J. Mol. Biol. 2014, 426, 2840–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef]
- Janini, M.; Rogers, M.; Birx, D.R.; McCutchan, F.E. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J. Virol. 2001, 75, 7973–7986. [Google Scholar] [CrossRef] [Green Version]
- Albin, J.S.; Harris, R.S. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: Implications for therapeutics. Expert Rev. Mol. Med. 2010, 12, e4. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Lorenzo-Redondo, R.; Little, S.J.; Chung, Y.S.; Phalora, P.K.; Maljkovic Berry, I.; Archer, J.; Penugonda, S.; Fischer, W.; Richman, D.D.; et al. Human APOBEC3 induced mutation of human immunodeficiency virus type-1 contributes to adaptation and evolution in natural infection. PLoS Pathog. 2014, 10, e1004281. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanini, F.; Puller, V.; Brodin, J.; Albert, J.; Neher, R.A. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus. Evol. 2017, 3, vex003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, K.D.; Monajemi, M.; Woodworth, C.F.; Grant, M.D.; Larijani, M. Impact of APOBEC Mutations on CD8+ T Cell Recognition of HIV Epitopes Varies Depending on the Restricting HLA. J. Acquir. Immune Defic. Syndr. 2015, 70, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Monajemi, M.; Woodworth, C.F.; Zipperlen, K.; Gallant, M.; Grant, M.D.; Larijani, M. Positioning of APOBEC3G/F mutational hotspots in the human immunodeficiency virus genome favors reduced recognition by CD8 + T cells. PLoS ONE 2014, 9, e93428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borzooee, F.; Joris, K.D.; Grant, M.D.; Larijani, M. APOBEC3G Regulation of the Evolutionary Race Between Adaptive Immunity and Viral Immune Escape Is Deeply Imprinted in the HIV Genome. Front. Immunol. 2018, 9, 3032. [Google Scholar] [CrossRef]
- Armitage, A.E.; Deforche, K.; Welch, J.J.; Van Laethem, K.; Camacho, R.; Rambaut, A.; Iversen, A.K. Possible footprints of APOBEC3F and/or other APOBEC3 deaminases, but not APOBEC3G, on HIV-1 from patients with acute/early and chronic infections. J. Virol. 2014, 88, 12882–12894. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Ooms, M.; Letko, M.; Garrett, N.; Simon, V.; Ndung’u, T. Functional characterization of Vif proteins from HIV-1 infected patients with different APOBEC3G haplotypes. AIDS 2016, 30, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Winkler, C.A.; Werner, L.; Mlisana, K.; Abdool Karim, S.S.; Ndung’u, T.; Caprisa Acute Infection Study Team. APOBEC3G expression is dysregulated in primary HIV-1 infection and polymorphic variants influence CD4+ T-cell counts and plasma viral load. AIDS 2010, 24, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Bunupuradah, T.; Imahashi, M.; Iampornsin, T.; Matsuoka, K.; Iwatani, Y.; Puthanakit, T.; Ananworanich, J.; Sophonphan, J.; Mahanontharit, A.; Naoe, T.; et al. Association of APOBEC3G genotypes and CD4 decline in Thai and Cambodian HIV-infected children with moderate immune deficiency. AIDS Res. Ther. 2012, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Bleiber, G.; Duggal, P.; Nelson, G.; May, M.; Mangeat, B.; Alobwede, I.; Trono, D.; Vlahov, D.; Donfield, S.; et al. APOBEC3G genetic variants and their influence on the progression to AIDS. J. Virol. 2004, 78, 11070–11076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.K.; Wang, Y.; Gray, K.P.; Farhad, M.; Brummel, S.; Fenton, T.; Trout, R.; Spector, S.A. Genetic variants in the host restriction factor APOBEC3G are associated with HIV-1-related disease progression and central nervous system impairment in children. J. Acquir. Immune Defic. Syndr. 2013, 62, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Lee, G.Q.; Reddy, K.; Einkauf, K.B.; Gounder, K.; Chevalier, J.M.; Dong, K.L.; Walker, B.D.; Yu, X.G.; Ndung’u, T.; Lichterfeld, M. HIV-1 DNA sequence diversity and evolution during acute subtype C infection. Nat. Commun. 2019, 10, 2737. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.G.; Ensoli, B.; Ivanoff, L.; Chamberlain, M.; Petteway, S.; Ratner, L.; Gallo, R.C.; Wong-Staal, F. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science 1987, 237, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, N.C.; Franchini, G.; Wong-Staal, F.; DuBois, G.C.; Robey, W.G.; Lautenberger, J.A.; Papas, T.S. Identification of HTLV-III/LAV sor gene product and detection of antibodies in human sera. Science 1986, 231, 1553–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Coligan, J.E.; Allan, J.S.; McLane, M.F.; Groopman, J.E.; Essex, M. A new HTLV-III/LAV protein encoded by a gene found in cytopathic retroviruses. Science 1986, 231, 1546–1549. [Google Scholar] [CrossRef]
- Strebel, K.; Daugherty, D.; Clouse, K.; Cohen, D.; Folks, T.; Martin, M.A. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 1987, 328, 728–730. [Google Scholar] [CrossRef]
- von Schwedler, U.; Song, J.; Aiken, C.; Trono, D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993, 67, 4945–4955. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 2013, 49, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhain, R.; Xu, J.; Burton, L.A.; Gordon, D.E.; Hultquist, J.F.; Johnson, J.R.; Satkamp, L.; Hiatt, J.; Rhee, D.Y.; Baek, K.; et al. ARIH2 Is a Vif-Dependent Regulator of CUL5-Mediated APOBEC3G Degradation in HIV Infection. Cell Host Microbe 2019, 26, 86–99.e87. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Akari, H.; Sakurai, A.; Yoshida, A.; Chiba, T.; Tanaka, K.; Strebel, K.; Adachi, A. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004, 6, 791–798. [Google Scholar] [CrossRef]
- Izumi, T.; Takaori-Kondo, A.; Shirakawa, K.; Higashitsuji, H.; Itoh, K.; Io, K.; Matsui, M.; Iwai, K.; Kondoh, H.; Sato, T.; et al. MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Shindo, K.; Nagata, K.; Yoshinaga, N.; Shirakawa, K.; Kobayashi, M.; Takaori-Kondo, A. Core Binding Factor beta Protects HIV, Type 1 Accessory Protein Viral Infectivity Factor from MDM2-mediated Degradation. J. Biol. Chem. 2016, 291, 24892–24899. [Google Scholar] [CrossRef] [Green Version]
- Albin, J.S.; Anderson, J.S.; Johnson, J.R.; Harjes, E.; Matsuo, H.; Krogan, N.J.; Harris, R.S. Dispersed sites of HIV Vif-dependent polyubiquitination in the DNA deaminase APOBEC3F. J. Mol. Biol. 2013, 425, 1172–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.; Shao, Q.; Wang, W.; Wang, Y.; Wang, C.; Kinlock, B.; Liu, B. Differential Contributions of Ubiquitin-Modified APOBEC3G Lysine Residues to HIV-1 Vif-Induced Degradation. J. Mol. Biol. 2016, 428, 3529–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Mercenne, G.; Bernacchi, S.; Richer, D.; Bec, G.; Henriet, S.; Paillart, J.C.; Marquet, R. HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 2010, 38, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.; Khan, M.A.; Miyagi, E.; Plishka, R.; Buckler-White, A.; Strebel, K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 2003, 77, 11398–11407. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, S.; Libre, C.; Batisse, J.; Mercenne, G.; Richer, D.; Laumond, G.; Decoville, T.; Moog, C.; Marquet, R.; Paillart, J.C. Translational regulation of APOBEC3G mRNA by Vif requires its 5′UTR and contributes to restoring HIV-1 infectivity. Sci. Rep. 2016, 6, 39507. [Google Scholar] [CrossRef]
- Pan, T.; Song, Z.; Wu, L.; Liu, G.; Ma, X.; Peng, Z.; Zhou, M.; Liang, L.; Liu, B.; Liu, J.; et al. USP49 potently stabilizes APOBEC3G protein by removing ubiquitin and inhibits HIV-1 replication. elife 2019, 8, e48318. [Google Scholar] [CrossRef]
- Miyakawa, K.; Matsunaga, S.; Kanou, K.; Matsuzawa, A.; Morishita, R.; Kudoh, A.; Shindo, K.; Yokoyama, M.; Sato, H.; Kimura, H.; et al. ASK1 restores the antiviral activity of APOBEC3G by disrupting HIV-1 Vif-mediated counteraction. Nat. Commun. 2015, 6, 6945. [Google Scholar] [CrossRef] [Green Version]
- Evilya, Z.; Chorin, E.; Gal-Garber, O.; Zelinger, E.; Turner, D.; Avidor, B.; Berke, G.; Hassin, D. Killing of Latently HIV-Infected CD4 T Cells by Autologous CD8 T Cells Is Modulated by Nef. Front. Immunol. 2018, 9, 2068. [Google Scholar] [CrossRef]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, T.; Chaudhary, P.; Gupta, K.; Islam, S.; Ghosh, P.; Santra, M.K.; Mitra, D. Cyclin F/FBXO1 Interacts with HIV-1 Viral Infectivity Factor (Vif) and Restricts Progeny Virion Infectivity by Ubiquitination and Proteasomal Degradation of Vif Protein through SCF(cyclin F) E3 Ligase Machinery. J. Biol. Chem. 2017, 292, 5349–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, R.; Takeuchi, J.S.; Yamada, E.; Nakano, Y.; Misawa, N.; Kimura, Y.; Ren, F.; Miyazawa, T.; Koyanagi, Y.; Sato, K. Feline Immunodeficiency Virus Evolutionarily Acquires Two Proteins, Vif and Protease, Capable of Antagonizing Feline APOBEC3. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Symeonides, M.; Albin, J.S.; Li, M.; Thali, M.; Harris, R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog. 2018, 14, e1007010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Wang, Y.; Tokunaga, K.; Huang, F.; Sun, B.; Yang, R. The HIV-1 accessory protein Vpr induces the degradation of the anti-HIV-1 agent APOBEC3G through a VprBP-mediated proteasomal pathway. Virus Res. 2015, 195, 25–34. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yodoi, J.; Sagawa, K.; Takatsuki, K.; Uchino, H. Adult T-cell leukemia: Clinical and hematologic features of 16 cases. Blood 1977, 50, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.P.; Matsuoka, M. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef] [Green Version]
- Mahieux, R.; Suspene, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef] [Green Version]
- Sasada, A.; Takaori-Kondo, A.; Shirakawa, K.; Kobayashi, M.; Abudu, A.; Hishizawa, M.; Imada, K.; Tanaka, Y.; Uchiyama, T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2005, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Ooms, M.; Krikoni, A.; Kress, A.K.; Simon, V.; Munk, C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012, 86, 6097–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Tanaka, M.; Takenouchi, N.; Ren, Y.; Lee, S.I.; Fujisawa, J.I. Induction of APOBEC3B cytidine deaminase in HTLV-1-infected humanized mice. Exp. Ther. Med. 2019, 17, 3701–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. BioMed Res. Int. 2013, 2013, 683095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Wattel, E.; Vartanian, J.P.; Pannetier, C.; Wain-Hobson, S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J. Virol. 1995, 69, 2863–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavrois, M.; Wain-Hobson, S.; Gessain, A.; Plumelle, Y.; Wattel, E. Adult T-cell leukemia/lymphoma on a background of clonally expanding human T-cell leukemia virus type-1-positive cells. Blood 1996, 88, 4646–4650. [Google Scholar] [CrossRef] [Green Version]
- Cavrois, M.; Leclercq, I.; Gout, O.; Gessain, A.; Wain-Hobson, S.; Wattel, E. Persistent oligoclonal expansion of human T-cell leukemia virus type 1-infected circulating cells in patients with Tropical spastic paraparesis/HTLV-1 associated myelopathy. Oncogene 1998, 17, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef]
- Singh, G.B.; Byun, H.; Ali, A.F.; Medina, F.; Wylie, D.; Shivram, H.; Nash, A.K.; Lozano, M.M.; Dudley, J.P. A Protein Antagonist of Activation-Induced Cytidine Deaminase Encoded by a Complex Mouse Retrovirus. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, F.; Lozano, M.; Dudley, J.P. C3H mouse mammary tumor virus superantigen function requires a splice donor site in the envelope gene. J. Virol. 2000, 74, 9431–9440. [Google Scholar] [CrossRef] [Green Version]
- Langlois, M.A.; Kemmerich, K.; Rada, C.; Neuberger, M.S. The AKV murine leukemia virus is restricted and hypermutated by mouse APOBEC3. J. Virol. 2009, 83, 11550–11559. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Stoye, J.P.; Coffin, J.M. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007, 3, 2014–2022. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Huang, F.T.; Lieber, M.R. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J. Biol. Chem. 2004, 279, 6496–6500. [Google Scholar] [CrossRef] [Green Version]
- Halemano, K.; Guo, K.; Heilman, K.J.; Barrett, B.S.; Smith, D.S.; Hasenkrug, K.J.; Santiago, M.L. Immunoglobulin somatic hypermutation by APOBEC3/Rfv3 during retroviral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7759–7764. [Google Scholar] [CrossRef] [Green Version]
- MacMillan, A.L.; Kohli, R.M.; Ross, S.R. APOBEC3 inhibition of mouse mammary tumor virus infection: The role of cytidine deamination versus inhibition of reverse transcription. J. Virol. 2013, 87, 4808–4817. [Google Scholar] [CrossRef] [Green Version]
- Rogozin, I.B.; Diaz, M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J. Immunol. 2004, 172, 3382–3384. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Chahwan, R.; Wang, S.; Wang, X.; Pham, P.T.; Goodman, M.F.; Bergman, A.; Scharff, M.D.; MacCarthy, T. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc. Natl. Acad. Sci. USA 2015, 112, E728–E737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, F.; Bhadra, S.; Johnston, D.; Lozano, M.; Dudley, J.P. The type B leukemogenic virus truncated superantigen is dispensable for T-cell lymphomagenesis. J. Virol. 2003, 77, 3866–3870. [Google Scholar] [CrossRef] [Green Version]
- Rosales Gerpe, M.C.; Renner, T.M.; Belanger, K.; Lam, C.; Aydin, H.; Langlois, M.A. N-linked glycosylation protects gammaretroviruses against deamination by APOBEC3 proteins. J. Virol. 2015, 89, 2342–2357. [Google Scholar] [CrossRef] [Green Version]
- Boi, S.; Ferrell, M.E.; Zhao, M.; Hasenkrug, K.J.; Evans, L.H. Mouse APOBEC3 expression in NIH 3T3 cells mediates hypermutation of AKV murine leukemia virus. Virology 2018, 518, 377–384. [Google Scholar] [CrossRef]
- Edwards, S.A.; Fan, H. gag-Related polyproteins of Moloney murine leukemia virus: Evidence for independent synthesis of glycosylated and unglycosylated forms. J. Virol. 1979, 30, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, L.H.; Dresler, S.; Kabat, D. Synthesis and glycosylation of polyprotein precursors to the internal core proteins of Friend murine leukemia virus. J. Virol. 1977, 24, 865–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polna, I.; Aleksandrowicz, J. Effect of adsorbents on IgM and IgG measles antibodies. Acta Virol. 1975, 19, 449–456. [Google Scholar] [PubMed]
- Kolokithas, A.; Rosenke, K.; Malik, F.; Hendrick, D.; Swanson, L.; Santiago, M.L.; Portis, J.L.; Hasenkrug, K.J.; Evans, L.H. The glycosylated Gag protein of a murine leukemia virus inhibits the antiretroviral function of APOBEC3. J. Virol. 2010, 84, 10933–10936. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, S.; Nitta, T.; Kotla, S.; Ha, D.; Nagashima, K.; Rein, A.R.; Fan, H.; Ross, S.R. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9078–9083. [Google Scholar] [CrossRef] [Green Version]
- Renner, T.M.; Belanger, K.; Lam, C.; Gerpe, M.C.R.; McBane, J.E.; Langlois, M.A. Full-Length Glycosylated Gag of Murine Leukemia Virus Can Associate with the Viral Envelope as a Type I Integral Membrane Protein. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Prats, A.C.; De Billy, G.; Wang, P.; Darlix, J.L. CUG initiation codon used for the synthesis of a cell surface antigen coded by the murine leukemia virus. J. Mol. Biol. 1989, 205, 363–372. [Google Scholar] [CrossRef]
- Edwards, S.A.; Fan, H. Sequence relationship of glycosylated and unglycosylated gag polyproteins of Moloney murine leukemia virus. J. Virol. 1980, 35, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, R.; McAtee, F.J.; Zirbel, J.H.; Portis, J.L. Characterization of glycosylated Gag expressed by a neurovirulent murine leukemia virus: Identification of differences in processing in vitro and in vivo. J. Virol. 1997, 71, 5355–5360. [Google Scholar] [CrossRef] [Green Version]
- Low, A.; Datta, S.; Kuznetsov, Y.; Jahid, S.; Kothari, N.; McPherson, A.; Fan, H. Mutation in the glycosylated gag protein of murine leukemia virus results in reduced in vivo infectivity and a novel defect in viral budding or release. J. Virol. 2007, 81, 3685–3692. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, R.; McAtee, F.J.; Favara, C.; Hayes, S.F.; Portis, J.L. N-terminal cleavage fragment of glycosylated Gag is incorporated into murine oncornavirus particles. J. Virol. 2001, 75, 11239–11243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, V.; Guetard, D.; Renard, M.; Keriel, A.; Sitbon, M.; Wain-Hobson, S.; Vartanian, J.P. Murine APOBEC1 is a powerful mutator of retroviral and cellular RNA in vitro and in vivo. J. Mol. Biol. 2009, 385, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Barrett, B.S.; Guo, K.; Harper, M.S.; Li, S.X.; Heilman, K.J.; Davidson, N.O.; Santiago, M.L. Reassessment of murine APOBEC1 as a retrovirus restriction factor in vivo. Virology 2014, 468–470, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Blanc, V.; Park, E.; Schaefer, S.; Miller, M.; Lin, Y.; Kennedy, S.; Billing, A.M.; Ben Hamidane, H.; Graumann, J.; Mortazavi, A.; et al. Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol. 2014, 15, R79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, V.; Davidson, N.O. APOBEC-1-mediated RNA editing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, T.; Kang, J. Immunological Genome Project and systems immunology. Trends Immunol. 2013, 34, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Lochelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [Green Version]
- Chareza, S.; Slavkovic Lukic, D.; Liu, Y.; Rathe, A.M.; Munk, C.; Zabogli, E.; Pistello, M.; Lochelt, M. Molecular and functional interactions of cat APOBEC3 and feline foamy and immunodeficiency virus proteins: Different ways to counteract host-encoded restriction. Virology 2012, 424, 138–146. [Google Scholar] [CrossRef]
- Jaguva Vasudevan, A.A.; Perkovic, M.; Bulliard, Y.; Cichutek, K.; Trono, D.; Haussinger, D.; Munk, C. Prototype foamy virus Bet impairs the dimerization and cytosolic solubility of human APOBEC3G. J. Virol. 2013, 87, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzer, W.M.; Tang, S.; Ahuka-Mundeke, S.; Shankar, A.; Hanson, D.L.; Zheng, H.; Ayouba, A.; Wolfe, N.D.; LeBreton, M.; Djoko, C.F.; et al. Novel simian foamy virus infections from multiple monkey species in women from the Democratic Republic of Congo. Retrovirology 2012, 9, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessain, A.; Rua, R.; Betsem, E.; Turpin, J.; Mahieux, R. HTLV-3/4 and simian foamy retroviruses in humans: Discovery, epidemiology, cross-species transmission and molecular virology. Virology 2013, 435, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boneva, R.S.; Switzer, W.M.; Spira, T.J.; Bhullar, V.B.; Shanmugam, V.; Cong, M.E.; Lam, L.; Heneine, W.; Folks, T.M.; Chapman, L.E. Clinical and virological characterization of persistent human infection with simian foamy viruses. AIDS Res. Hum. Retrovir. 2007, 23, 1330–1337. [Google Scholar] [CrossRef]
- Betsem, E.; Rua, R.; Tortevoye, P.; Froment, A.; Gessain, A. Frequent and recent human acquisition of simian foamy viruses through apes’ bites in central Africa. PLoS Pathog. 2011, 7, e1002306. [Google Scholar] [CrossRef]
- Matsen, F.A.t.; Small, C.T.; Soliven, K.; Engel, G.A.; Feeroz, M.M.; Wang, X.; Craig, K.L.; Hasan, M.K.; Emerman, M.; Linial, M.L.; et al. A novel Bayesian method for detection of APOBEC3-mediated hypermutation and its application to zoonotic transmission of simian foamy viruses. PLoS Comput. Biol. 2014, 10, e1003493. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, M.; Li, Y.; Li, K.; Wu, S.; Guo, T.; Cen, S.; Jiang, J.; Li, Z.; Li, Y. APOBEC3G is a restriction factor of EV71 and mediator of IMB-Z antiviral activity. Antivir. Res. 2019, 165, 23–33. [Google Scholar] [CrossRef]
- Tiwarekar, V.; Wohlfahrt, J.; Fehrholz, M.; Scholz, C.J.; Kneitz, S.; Schneider-Schaulies, J. APOBEC3G-Regulated Host Factors Interfere with Measles Virus Replication: Role of REDD1 and Mammalian TORC1 Inhibition. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Li, Z.; Ning, S.; Su, X.; Liu, X.; Wang, H.; Liu, Y.; Zheng, W.; Zheng, B.; Yu, X.F.; Zhang, W. Enterovirus 71 antagonizes the inhibition of the host intrinsic antiviral factor A3G. Nucleic Acids Res. 2018, 46, 11514–11527. [Google Scholar] [CrossRef]
- Fehrholz, M.; Kendl, S.; Prifert, C.; Weissbrich, B.; Lemon, K.; Rennick, L.; Duprex, P.W.; Rima, B.K.; Koning, F.A.; Holmes, R.K.; et al. The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J. Gen. Virol. 2012, 93, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Malim, M.H. APOBEC-mediated editing of viral RNA. Science 2004, 305, 645. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Sure, K.; Ihorst, G.; Stang, A.; Pyrc, K.; Jebbink, M.F.; Petersen, G.; Forster, J.; Berkhout, B.; Uberla, K. Croup is associated with the novel coronavirus NL63. PLoS Med. 2005, 2, e240. [Google Scholar] [CrossRef]
- Pyrc, K.; Berkhout, B.; van der Hoek, L. The novel human coronaviruses NL63 and HKU1. J. Virol. 2007, 81, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Wang, S.M.; Wang, C.T. APOBEC3G cytidine deaminase association with coronavirus nucleocapsid protein. Virology 2009, 388, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Hozumi, Y.; Zheng, Y.H.; Yin, C.; Wei, G.W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses 2020, 12, 1095. [Google Scholar] [CrossRef]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef]
- Herbert, A. ADAR and Immune Silencing in Cancer. Trends Cancer 2019, 5, 272–282. [Google Scholar] [CrossRef]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.T.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekerman, E.; Jeon, D.; Ardolino, M.; Coscoy, L. A role for host activation-induced cytidine deaminase in innate immune defense against KSHV. PLoS Pathog. 2013, 9, e1003748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrovnitchaia, I.; Valieris, R.; Drummond, R.D.; Lima, J.P.; Freitas, H.C.; Bartelli, T.F.; de Amorim, M.G.; Nunes, D.N.; Dias-Neto, E.; da Silva, I.T. APOBEC-mediated DNA alterations: A possible new mechanism of carcinogenesis in EBV-positive gastric cancer. Int. J. Cancer 2020, 146, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E.; et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 2019, 4, 78–88. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Attarian, C.; Yockteng-Melgar, J.; Jarvis, M.C.; Biolatti, M.; Galitska, G.; Dell’Oste, V.; Frappier, L.; Bierle, C.J.; et al. A Conserved Mechanism of APOBEC3 Relocalization by Herpesviral Ribonucleotide Reductase Large Subunits. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Sakowski, E.G.; Munsell, E.V.; Hyatt, M.; Kress, W.; Williamson, S.J.; Nasko, D.J.; Polson, S.W.; Wommack, K.E. Ribonucleotide reductases reveal novel viral diversity and predict biological and ecological features of unknown marine viruses. Proc. Natl. Acad. Sci. USA 2014, 111, 15786–15791. [Google Scholar] [CrossRef] [Green Version]
- Torrents, E. Ribonucleotide reductases: Essential enzymes for bacterial life. Front. Cell. Infect. Microbiol. 2014, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Pereira, L.; Maidji, E.; McDonagh, S.; Tabata, T. Insights into viral transmission at the uterine-placental interface. Trends Microbiol. 2005, 13, 164–174. [Google Scholar] [CrossRef]
- MacLachlan, J.H.; Cowie, B.C. Hepatitis B virus epidemiology. Cold Spring Harb. Perspect. Med. 2015, 5, a021410. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.A.; Hu, J. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J. Virol. 2013, 87, 2563–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Zlotnick, A. Asymmetric Modification of Hepatitis B Virus (HBV) Genomes by an Endogenous Cytidine Deaminase inside HBV Cores Informs a Model of Reverse Transcription. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kock, J.; Blum, H.E. Hypermutation of hepatitis B virus genomes by APOBEC3G, APOBEC3C and APOBEC3H. J. Gen. Virol. 2008, 89, 1184–1191. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hu, J. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 2008, 82, 6852–6861. [Google Scholar] [CrossRef] [Green Version]
- Bockmann, J.H.; Stadler, D.; Xia, Y.; Ko, C.; Wettengel, J.M.; Schulze Zur Wiesch, J.; Dandri, M.; Protzer, U. Comparative Analysis of the Antiviral Effects Mediated by Type I and III Interferons in Hepatitis B Virus-Infected Hepatocytes. J. Infect. Dis. 2019, 220, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Eggerman, T.L.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Kurlander, R.; Patterson, A.P. Heat shock proteins stimulate APOBEC-3-mediated cytidine deamination in the hepatitis B virus. J. Biol. Chem. 2017, 292, 13459–13479. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhao, X.; Wang, Y.; Xie, Y.; Liu, J. Hepatitis B virus X protein is capable of down-regulating protein level of host antiviral protein APOBEC3G. Sci. Rep. 2017, 7, 40783. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhang, X.; Ding, H.; Qiao, Y.; Han, X.; Geng, W.; Guan, G.; Cui, H.; Zhao, B.; Wu, Y.; et al. AID recruits the RNA exosome to degrade HIV-1 nascent transcripts through interaction with the Tat-P-TEFb-TAR RNP complex. FEBS Lett. 2018, 592, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Takeuchi, H.; Strebel, K.; Levin, J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J. Virol. 2006, 80, 5992–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, M.B.; Webb, J.; Chelico, L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PLoS ONE 2013, 8, e64196. [Google Scholar] [CrossRef] [Green Version]
- Belanger, K.; Savoie, M.; Rosales Gerpe, M.C.; Couture, J.F.; Langlois, M.A. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 2013, 41, 7438–7452. [Google Scholar] [CrossRef]
- Chaurasiya, K.R.; McCauley, M.J.; Wang, W.; Qualley, D.F.; Wu, T.; Kitamura, S.; Geertsema, H.; Chan, D.S.; Hertz, A.; Iwatani, Y.; et al. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014, 6, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.; Huo, R.; Feng, Y.; Rouzina, I.; Chelico, L.; Williams, M.C. Dimerization regulates both deaminase-dependent and deaminase-independent HIV-1 restriction by APOBEC3G. Nat. Commun. 2017, 8, 597. [Google Scholar] [CrossRef] [Green Version]
- Browne, E.P.; Allers, C.; Landau, N.R. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 2009, 387, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, E.; Opi, S.; Takeuchi, H.; Khan, M.; Goila-Gaur, R.; Kao, S.; Strebel, K. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 2007, 81, 13346–13353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, A.J.; Hache, G.; Macduff, D.A.; Brown, W.L.; Harris, R.S. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 2008, 82, 2652–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieniasz, P.D. A multimodal antiretroviral protein. Nat. Microbiol. 2018, 3, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Adolph, M.B.; Ara, A.; Chelico, L. APOBEC3 Host Restriction Factors of HIV-1 Can Change the Template Switching Frequency of Reverse Transcriptase. J. Mol. Biol. 2019, 431, 1339–1352. [Google Scholar] [CrossRef]
- Temin, H.M. Retrovirus variation and reverse transcription: Abnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA 1993, 90, 6900–6903. [Google Scholar] [CrossRef] [Green Version]
- Basu, V.P.; Song, M.; Gao, L.; Rigby, S.T.; Hanson, M.N.; Bambara, R.A. Strand transfer events during HIV-1 reverse transcription. Virus. Res. 2008, 134, 19–38. [Google Scholar] [CrossRef]
- Johnson, P.E.; Turner, R.B.; Wu, Z.R.; Hairston, L.; Guo, J.; Levin, J.G.; Summers, M.F. A mechanism for plus-strand transfer enhancement by the HIV-1 nucleocapsid protein during reverse transcription. Biochemistry 2000, 39, 9084–9091. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Rawson, J.M.O.; Nikolaitchik, O.A.; Keele, B.F.; Pathak, V.K.; Hu, W.S. Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res. 2018, 46, 10535–10545. [Google Scholar] [CrossRef] [Green Version]
- Marino, D.; Perkovic, M.; Hain, A.; Jaguva Vasudevan, A.A.; Hofmann, H.; Hanschmann, K.M.; Muhlebach, M.D.; Schumann, G.G.; Konig, R.; Cichutek, K.; et al. APOBEC4 Enhances the Replication of HIV-1. PLoS ONE 2016, 11, e0155422. [Google Scholar] [CrossRef] [Green Version]
- Riquet, A.; Chevalier, S.; Villaudy, J.; Gazzolo, L.; Vartanian, J.-P.; Mahieux, R.; Duc-Dodon, M.; Bonnefoy, N. Human T-cell leukemia virus type 1 (HTLV-1) Tax oncoprotein induces DNA damages through Activation-Induced cytidine Deaminase (AID). Retrovirology 2014, 11, O45. [Google Scholar] [CrossRef] [Green Version]
- Okeoma, C.M.; Petersen, J.; Ross, S.R. Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J. Virol. 2009, 83, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, B.; Kraase, M.; Indikova, I.; Indik, S. A high rate of polymerization during synthesis of mouse mammary tumor virus DNA alleviates hypermutation by APOBEC3 proteins. PLoS Pathog. 2019, 15, e1007533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmons, B.; Miethke, T.; Wintersperger, S.; Muller, M.; Brem, G.; Gunzburg, W.H. Superantigen expression is driven by both mouse mammary tumor virus long terminal repeat-associated promoters in transgenic mice. J. Virol. 2000, 74, 2900–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, J.A.; Simper, M.S.; Lozano, M.M.; Payne, S.M.; Dudley, J.P. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J. Virol. 2005, 79, 14737–14747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, J.A.; Chadee, A.B.; Byun, H.; Russell, R.; Dudley, J.P. Mapping of the functional boundaries and secondary structure of the mouse mammary tumor virus Rem-responsive element. J. Biol. Chem. 2009, 284, 25642–25652. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.; Halani, N.; Gou, Y.; Nash, A.K.; Lozano, M.M.; Dudley, J.P. Requirements for mouse mammary tumor virus Rem signal peptide processing and function. J. Virol. 2012, 86, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Doehle, B.P.; Bogerd, H.P.; Wiegand, H.L.; Jouvenet, N.; Bieniasz, P.D.; Hunter, E.; Cullen, B.R. The betaretrovirus Mason-Pfizer monkey virus selectively excludes simian APOBEC3G from virion particles. J. Virol. 2006, 80, 12102–12108. [Google Scholar] [CrossRef] [Green Version]
- Low, A.; Okeoma, C.M.; Lovsin, N.; de las Heras, M.; Taylor, T.H.; Peterlin, B.M.; Ross, S.R.; Fan, H. Enhanced replication and pathogenesis of Moloney murine leukemia virus in mice defective in the murine APOBEC3 gene. Virology 2009, 385, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, S.; Crawford, D.; Blouch, K.; Browne, E.P.; Kohli, R.M.; Ross, S.R. Different modes of retrovirus restriction by human APOBEC3A and APOBEC3G in vivo. PLoS Pathog. 2014, 10, e1004145. [Google Scholar] [CrossRef]
- Browne, E.P.; Littman, D.R. Species-specific restriction of apobec3-mediated hypermutation. J. Virol. 2008, 82, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rulli, S.J., Jr.; Mirro, J.; Hill, S.A.; Lloyd, P.; Gorelick, R.J.; Coffin, J.M.; Derse, D.; Rein, A. Interactions of murine APOBEC3 and human APOBEC3G with murine leukemia viruses. J. Virol. 2008, 82, 6566–6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boi, S.; Kolokithas, A.; Shepard, J.; Linwood, R.; Rosenke, K.; Van Dis, E.; Malik, F.; Evans, L.H. Incorporation of mouse APOBEC3 into murine leukemia virus virions decreases the activity and fidelity of reverse transcriptase. J. Virol. 2014, 88, 7659–7662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrou, S.; Zhao, W.; Blouch, K.; Ross, S.R. Deaminase-Dead Mouse APOBEC3 Is an In Vivo Retroviral Restriction Factor. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Akkawi, C.; Mougel, M.; Ross, S.R. Murine Leukemia Virus P50 Protein Counteracts APOBEC3 by Blocking Its Packaging. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, J.; Bompard-Marechal, G.; Audit, M.; Hope, T.J.; Sitbon, M.; Mougel, M. A novel subgenomic murine leukemia virus RNA transcript results from alternative splicing. J. Virol. 2000, 74, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourzi, P.; Leonova, T.; Papavasiliou, F.N. Viral induction of AID is independent of the interferon and the Toll-like receptor signaling pathways but requires NF-kappaB. J. Exp. Med. 2007, 204, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourzi, P.; Leonova, T.; Papavasiliou, F.N. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity 2006, 24, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Ju, Y.; Chen, M.; Xie, Z.; Zhou, K.; Tan, Y.; Mo, J. Epidemiological and genetic characteristics of EV71 in hand, foot, and mouth disease in Guangxi, southern China, from 2010 to 2015. PLoS ONE 2017, 12, e0188640. [Google Scholar] [CrossRef] [Green Version]
- Cen, S.; Peng, Z.G.; Li, X.Y.; Li, Z.R.; Ma, J.; Wang, Y.M.; Fan, B.; You, X.F.; Wang, Y.P.; Liu, F.; et al. Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 2010, 285, 16546–16552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.Y.; Wang, H.Q.; Hao, L.H.; He, W.Y.; Gao, R.M.; Li, Y.P.; Li, Y.H.; Jiang, J.D.; Li, Z.R. Synthesis and antiviral activity of N-phenylbenzamide derivatives, a novel class of enterovirus 71 inhibitors. Molecules 2013, 18, 3630–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, E.K.; Schmolke, M.; Hofmann, H.; Ehrhardt, C.; Flory, E.; Munk, C.; Ludwig, S. High level expression of the anti-retroviral protein APOBEC3G is induced by influenza A virus but does not confer antiviral activity. Retrovirology 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Cao, R.; Huang, Y.; Zhou, H.; Liu, Y.; Li, X.; Zhong, W.; Hao, P. A comprehensive study on cellular RNA editing activity in response to infections with different subtypes of influenza a viruses. BMC Genom. 2018, 19, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, Y.; Stavrou, S.; Blouch, K.; Tattersall, P.; Ross, S.R. In Vivo Examination of Mouse APOBEC3- and Human APOBEC3A- and APOBEC3G-Mediated Restriction of Parvovirus and Herpesvirus Infection in Mouse Models. J. Virol. 2016, 90, 8005–8012. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lilley, C.E.; Yu, Q.; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLoS Pathog. 2020, 16, e1008718. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, J.; Wu, J.; Zhang, M.; Gao, P. Associations between activation-induced cytidine deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like cytidine deaminase expression, hepatitis B virus (HBV) replication and HBV-associated liver disease (Review). Mol. Med. Rep. 2015, 12, 6405–6414. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [Green Version]
- Seppen, J. Unedited inhibition of HBV replication by APOBEC3G. J. Hepatol 2004, 41, 1068–1069. [Google Scholar] [CrossRef]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [Green Version]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Henry, M.; Marchio, A.; Suspene, R.; Aynaud, M.M.; Guetard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J. Virol. 2016, 90, 6379–6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starrett, G.J.; Serebrenik, A.A.; Roelofs, P.A.; McCann, J.L.; Verhalen, B.; Jarvis, M.C.; Stewart, T.A.; Law, E.K.; Krupp, A.; Jiang, M.; et al. Polyomavirus T Antigen Induces APOBEC3B Expression Using an LXCXE-Dependent and TP53-Independent Mechanism. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, A.J.; Nissley, D.V.; Harris, R.S. APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc. Natl. Acad. Sci. USA 2005, 102, 9854–9859. [Google Scholar] [CrossRef] [Green Version]
- Niewiadomska, A.M.; Tian, C.; Tan, L.; Wang, T.; Sarkis, P.T.; Yu, X.F. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 2007, 81, 9577–9583. [Google Scholar] [CrossRef] [Green Version]
- Metzner, M.; Jack, H.M.; Wabl, M. LINE-1 retroelements complexed and inhibited by activation induced cytidine deaminase. PLoS ONE 2012, 7, e49358. [Google Scholar] [CrossRef] [Green Version]
- MacDuff, D.A.; Demorest, Z.L.; Harris, R.S. AID can restrict L1 retrotransposition suggesting a dual role in innate and adaptive immunity. Nucleic Acids Res. 2009, 37, 1854–1867. [Google Scholar] [CrossRef] [Green Version]
- Koito, A.; Ikeda, T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Knisbacher, B.A.; Levanon, E.Y. DNA and RNA editing of retrotransposons accelerate mammalian genome evolution. Ann. N. Y. Acad. Sci. 2015, 1341, 115–125. [Google Scholar] [CrossRef]
- Knisbacher, B.A.; Gerber, D.; Levanon, E.Y. DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet. 2016, 32, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Kinomoto, M.; Kanno, T.; Shimura, M.; Ishizaka, Y.; Kojima, A.; Kurata, T.; Sata, T.; Tokunaga, K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007, 35, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Heidmann, O.; Delebecque, F.; Dewannieux, M.; Ribet, D.; Hance, A.J.; Heidmann, T.; Schwartz, O. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature 2005, 433, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Dutko, J.A.; Schafer, A.; Kenny, A.E.; Cullen, B.R.; Curcio, M.J. Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr. Biol. 2005, 15, 661–666. [Google Scholar] [CrossRef] [Green Version]
- Adolph, M.B.; Love, R.P.; Chelico, L. Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infect. Dis. 2018, 4, 224–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Sequencing, C.; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA 2004, 101 (Suppl. 2), 14572–14579. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.N.; Malim, M.H.; Bieniasz, P.D. Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 2008, 82, 8762–8770. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Shimoda, M.; Ebrahimi, D.; VandeBerg, J.L.; Harris, R.S.; Koito, A.; Maeda, K. Opossum APOBEC1 is a DNA mutator with retrovirus and retroelement restriction activity. Sci. Rep. 2017, 7, 46719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treger, R.S.; Tokuyama, M.; Dong, H.; Salas-Briceno, K.; Ross, S.R.; Kong, Y.; Iwasaki, A. Human APOBEC3G Prevents Emergence of Infectious Endogenous Retrovirus in Mice. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knisbacher, B.A.; Levanon, E.Y. DNA Editing of LTR Retrotransposons Reveals the Impact of APOBECs on Vertebrate Genomes. Mol. Biol. Evol. 2016, 33, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, J.G.; Cristofari, G. Post-Transcriptional Control of LINE-1 Retrotransposition by Cellular Host Factors in Somatic Cells. Front. Cell Dev. Biol. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orecchini, E.; Frassinelli, L.; Galardi, S.; Ciafre, S.A.; Michienzi, A. Post-transcriptional regulation of LINE-1 retrotransposition by AID/APOBEC and ADAR deaminases. Chromosome Res. 2018, 26, 45–59. [Google Scholar] [CrossRef]
- Bogerd, H.P.; Wiegand, H.L.; Hulme, A.E.; Garcia-Perez, J.L.; O’Shea, K.S.; Moran, J.V.; Cullen, B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Muckenfuss, H.; Hamdorf, M.; Held, U.; Perkovic, M.; Lower, J.; Cichutek, K.; Flory, E.; Schumann, G.G.; Munk, C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 2006, 281, 22161–22172. [Google Scholar] [CrossRef] [Green Version]
- Stenglein, M.D.; Harris, R.S. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 2006, 281, 16837–16841. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.R.; Narvaiza, I.; Planegger, R.A.; Weitzman, M.D.; Moran, J.V. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. elife 2014, 3, e02008. [Google Scholar] [CrossRef]
- Horn, A.V.; Klawitter, S.; Held, U.; Berger, A.; Vasudevan, A.A.; Bock, A.; Hofmann, H.; Hanschmann, K.M.; Trosemeier, J.H.; Flory, E.; et al. Human LINE-1 restriction by APOBEC3C is deaminase independent and mediated by an ORF1p interaction that affects LINE reverse transcriptase activity. Nucleic Acids Res. 2014, 42, 396–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, T.; Arias, J.F.; Iwabu, Y.; Yokoyama, M.; Fujita, H.; Sato, H.; Tokunaga, K. APOBEC3G oligomerization is associated with the inhibition of both Alu and LINE-1 retrotransposition. PLoS ONE 2013, 8, e84228. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Xu, J.; Yuan, W.; Song, X.; Zhang, J.; Wei, W.; Yu, X.F.; Yang, Y. APOBEC3DE Inhibits LINE-1 Retrotransposition by Interacting with ORF1p and Influencing LINE Reverse Transcriptase Activity. PLoS ONE 2016, 11, e0157220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Goubran, M.H.; Follack, T.B.; Chelico, L. Deamination-independent restriction of LINE-1 retrotransposition by APOBEC3H. Sci. Rep. 2017, 7, 10881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Abd El Galil, K.H.; Tokunaga, K.; Maeda, K.; Sata, T.; Sakaguchi, N.; Heidmann, T.; Koito, A. Intrinsic restriction activity by apolipoprotein B mRNA editing enzyme APOBEC1 against the mobility of autonomous retrotransposons. Nucleic Acids Res. 2011, 39, 5538–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Sarkis, P.T.; Wang, T.; Tian, C.; Yu, X.F. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 2009, 23, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Wissing, S.; Montano, M.; Garcia-Perez, J.L.; Moran, J.V.; Greene, W.C. Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J. Biol. Chem. 2011, 286, 36427–36437. [Google Scholar] [CrossRef] [Green Version]
- Turelli, P.; Vianin, S.; Trono, D. The innate antiretroviral factor APOBEC3G does not affect human LINE-1 retrotransposition in a cell culture assay. J. Biol. Chem. 2004, 279, 43371–43373. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; MacCarthy, T. The preferred nucleotide contexts of the AID/APOBEC cytidine deaminases have differential effects when mutating retrotransposon and virus sequences compared to host genes. PLoS Comput. Biol. 2017, 13, e1005471. [Google Scholar] [CrossRef]
- Jung, H.; Choi, J.K.; Lee, E.A. Immune signatures correlate with L1 retrotransposition in gastrointestinal cancers. Genome Res. 2018, 28, 1136–1146. [Google Scholar] [CrossRef] [Green Version]




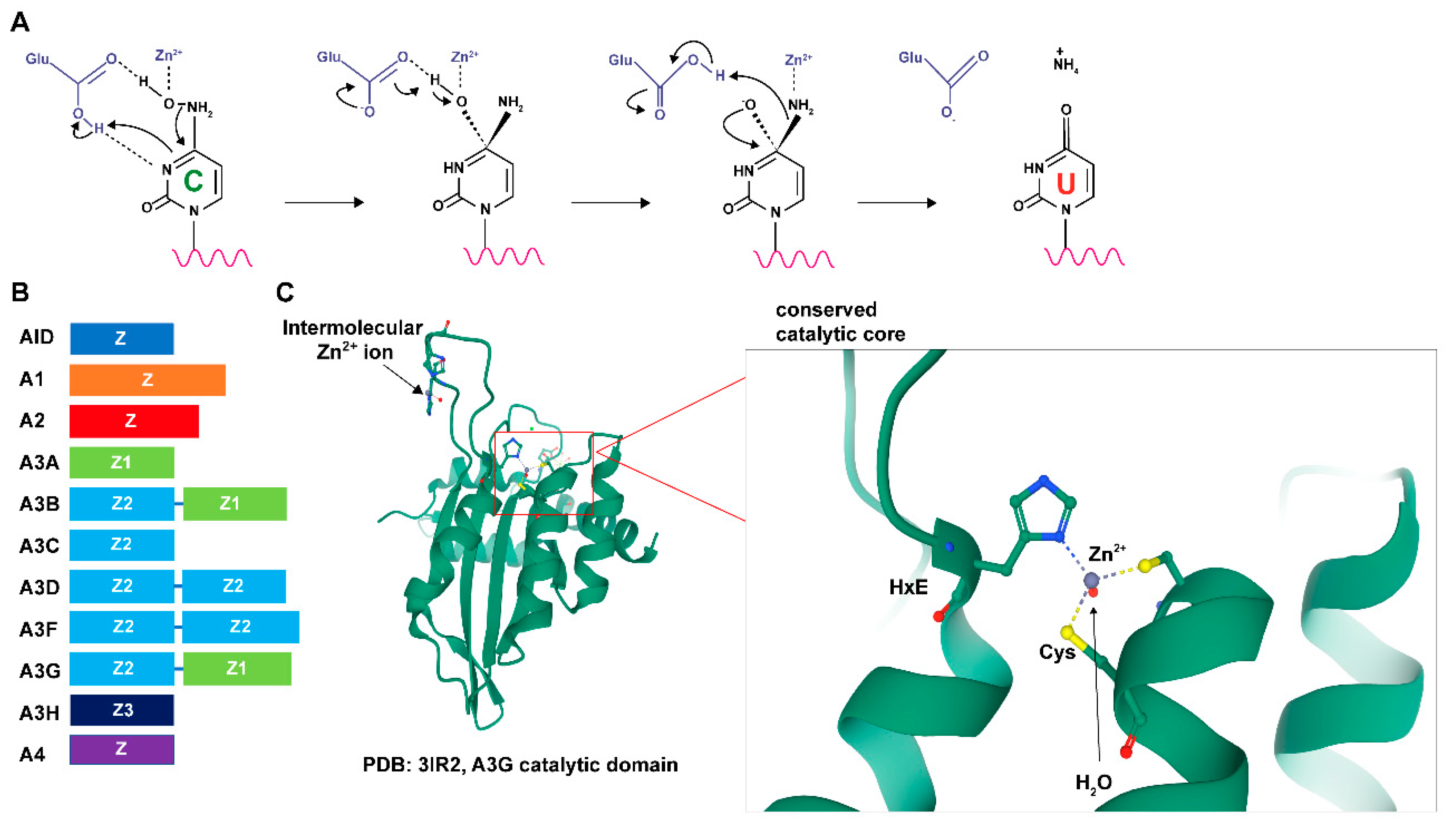{kind=link}
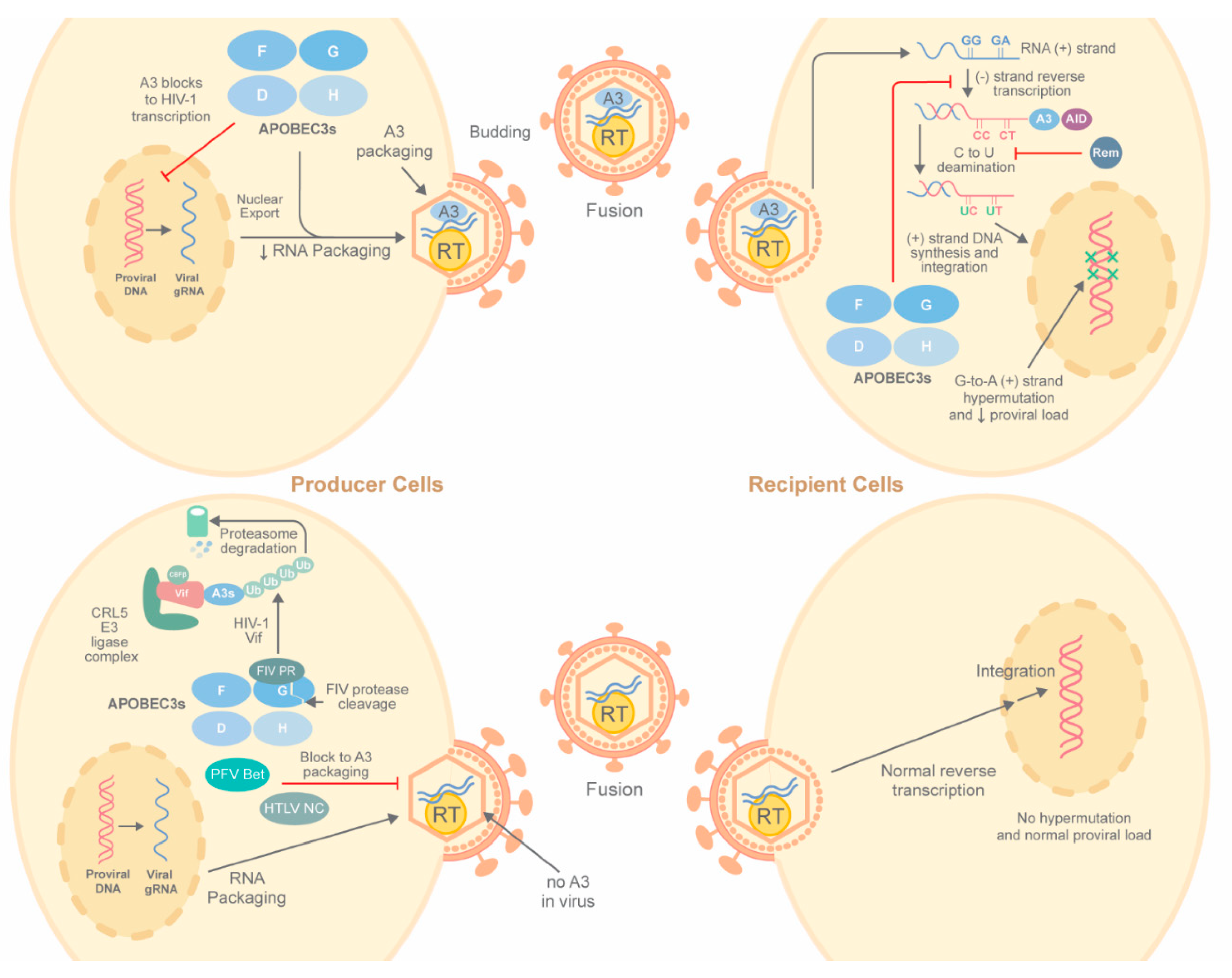{kind=link}
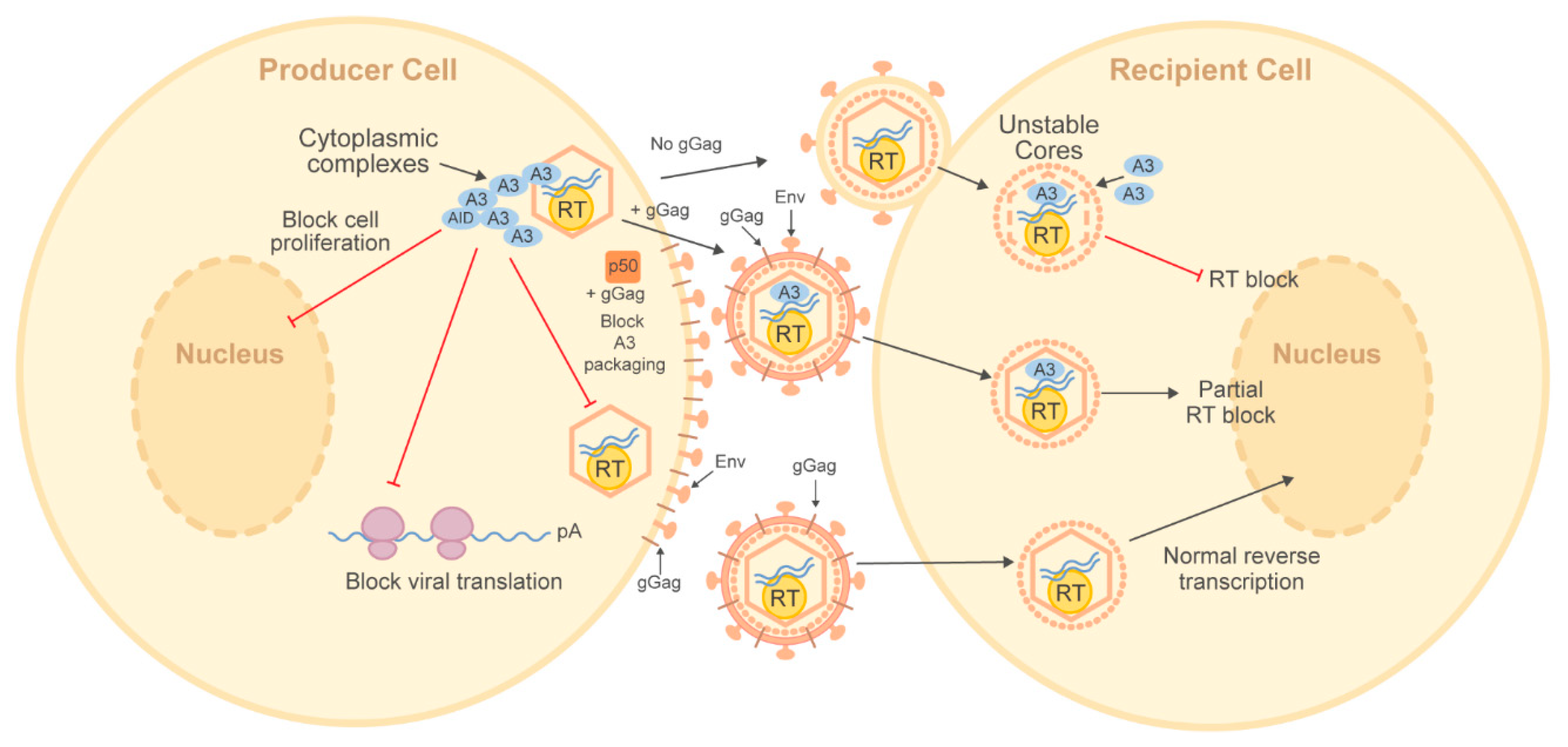{kind=link}
| Viral Family | Virus | APOBEC Protein | Mechanism of APOBEC Action | Viral APOBEC Antagonist | Mechanism of Viral Antagonist |
|---|---|---|---|---|---|
| Coronaviridae | SARS CoV-1/CoV-2 | hA1, hA3 | Hypermutation of viral RNA?? | Nucleocapsid?? | ?? |
| Hepadnaviridae | HBV | hA3 | Hypermutation; reduced viral DNA | HBx | Decreased hA3 levels by enhancing exocytosis |
| Herpesviridae | EBV | hA3 | Hypermutation; reduced viral DNA | BORF2 | Inhibited deaminase activity; altered A3 subcellular localization |
| Herpesviridae | HCMV | hA3 | Hypermutation; reduced viral DNA | ?? | ?? |
| Herpesviridae | HSV-1 | hA3 | Reduced viral DNA? | ICP6?? | Altered hA3 subcellular localization |
| Herpesviridae | KSHV | hA3 | Hypermutation; reduced viral DNA | ORF61 | Altered hA3 subcellular localization |
| Herpesviridae | KSHV | hAID 2 | Induced NKG2D ligands on NK cells | Viral miRs | Decreased hAID protein |
| Paramyxoviridae | MV | hA3 | Blocked translation of viral mRNA | ?? | ?? |
| Paramyxoviridae | NDV | chA4 2 | Reduced viral RNA | ?? | ?? |
| Parvoviridae | MVM | hA3 | Reduced viral DNA | ?? | ?? |
| Picornaviridae | EV71 | hA3 | Inhibited viral RNA synthesis and translation | 2C helicase; 3D RNA-dependent RNA polymerase | Autophagy-lysosomal degradation of A3G by 2C; reduced virion packaging by 3D?? |
| Retroviridae | Ab-MLV | mAID | Inhibited proliferation of infected cells; induced NKG2D ligands on NK cells | ?? | ?? |
| Retroviridae | AKV | mA3 | Hypermutation; reduced viral DNA | gGag | ?? |
| Retroviridae | FFV | fA3 | Hypermutation; reduced viral DNA | Bet | Sequestered fA3 reduces virion packaging |
| Retroviridae | FIV | fA3 2 | Hypermutation; reduced viral DNA | Vif; PR | fA3 degradation and cleavage |
| Retroviridae | HIV-1 | hAID | Inhibition of viral transcription | ?? | ?? |
| Retroviridae | HIV-1 SIV | hA3/ sA3 2,3 | Hypermutation, reduced transcription, increased defective proviruses, reduced viral DNA synthesis by binding RT; mutated viral epitopes for CTLs | Vif; Env; Vpr | hA3 degradation and reduced virion packaging; decreased A3 translation; increased Pol packaging |
| Retroviridae | HTLV-1 | hA3 | Hypermutation; inactivation of Tax | Nucleocapsid | Reduced hA3 packaging into virions; oligoclonal expansion of infected cells |
| Retroviridae | M-MLV | mA3 | Reduced viral DNA by binding RT | gGag | Unstable cores |
| Retroviridae | M-MLV, F-MLV | mA3 | Reduced viral DNA | p50 | Reduced mA3 packaging into virions |
| Retroviridae | MMTV | mAID 2 | Hypermutation; reduced proviral DNA | Rem | mAID degradation |
| Retroviridae | MMTV | mA3 | Reduced viral DNA | RT | Rapid polymerization |
| Retroviridae | MMTV | mA3 | Hypermutation?? | Rem | ?? |
| Retroviridae | MPMV | rA3 2 | ?? | ?? | Reduced rA3 packaging into virions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.K.; Byun, H.; Dudley, J.P. The Role of APOBECs in Viral Replication. Microorganisms 2020, 8, 1899. https://doi.org/10.3390/microorganisms8121899
Xu WK, Byun H, Dudley JP. The Role of APOBECs in Viral Replication. Microorganisms. 2020; 8(12):1899. https://doi.org/10.3390/microorganisms8121899
Chicago/Turabian StyleXu, Wendy Kaichun, Hyewon Byun, and Jaquelin P. Dudley. 2020. "The Role of APOBECs in Viral Replication" Microorganisms 8, no. 12: 1899. https://doi.org/10.3390/microorganisms8121899
APA StyleXu, W. K., Byun, H., & Dudley, J. P. (2020). The Role of APOBECs in Viral Replication. Microorganisms, 8(12), 1899. https://doi.org/10.3390/microorganisms8121899