The Nature and Extent of Plasmid Variation in Chlamydia trachomatis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction and Sequencing
2.3. SNP Detection
2.4. dN/dS Ratio
2.5. Maximum Distances between Plasmid Sequences
2.6. Variations in Number of 22 bp Repeats
2.7. Clustering and Rarefaction Analysis of Plasmid Sequences
2.8. Phylogenetic Reconstruction
3. Results
3.1. Overall Plasmid Diversity
3.2. Evolutionary Distances
3.3. dN/dS Ratio
3.4. SNP Characteristics
3.4.1. Position of SNP within the Codon
3.4.2. Ratio of Synonymous to Nonsynonymous SNPs
3.4.3. Amino Acid Substitution Characteristics
3.4.4. Occurrence of tri-allelic SNPs
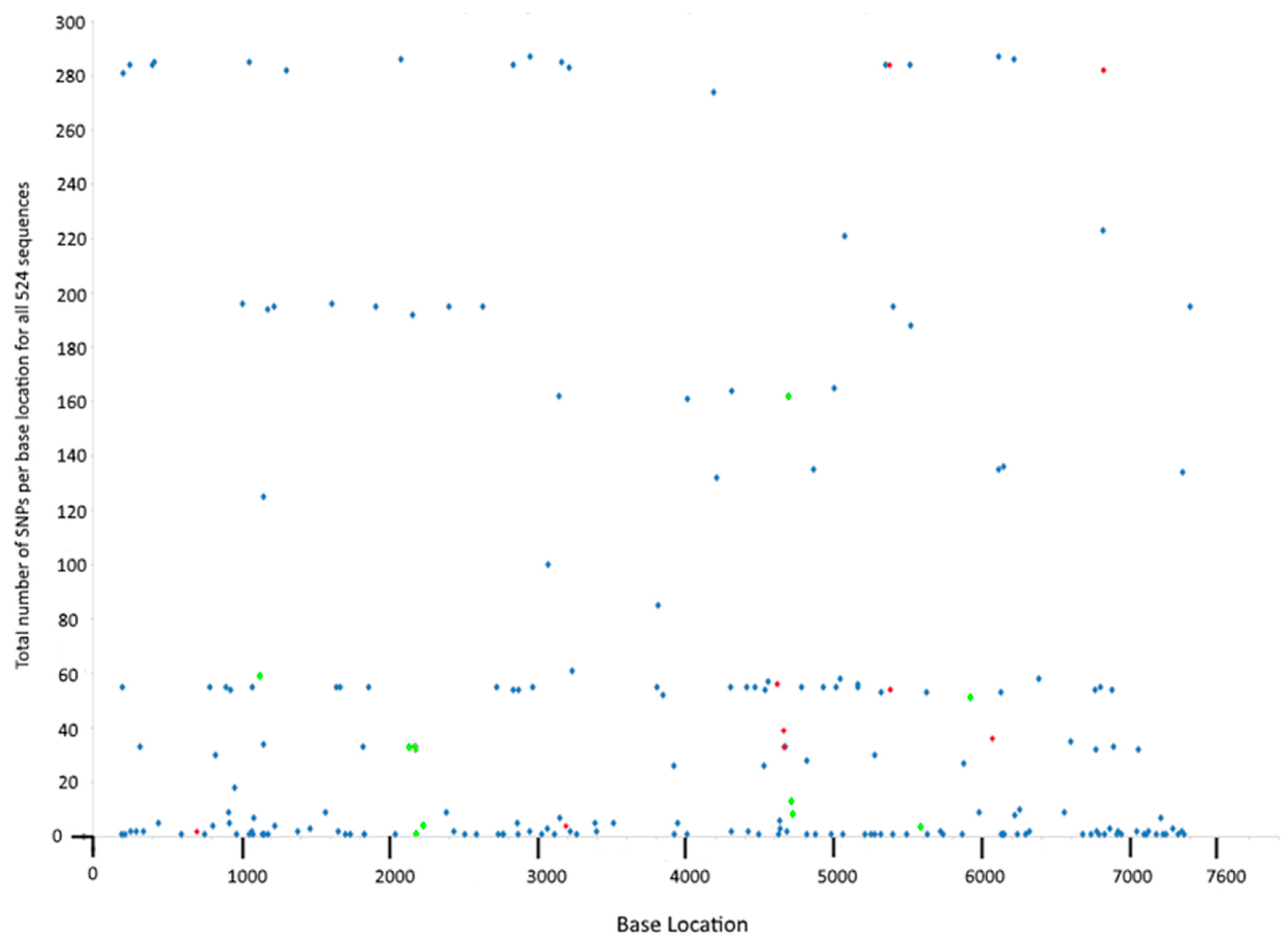
3.5. SNP Frequency at Specific Loci
3.6. Identification of Premature and Delayed Stop Codons
3.7. Phylogenetic Reconstruction
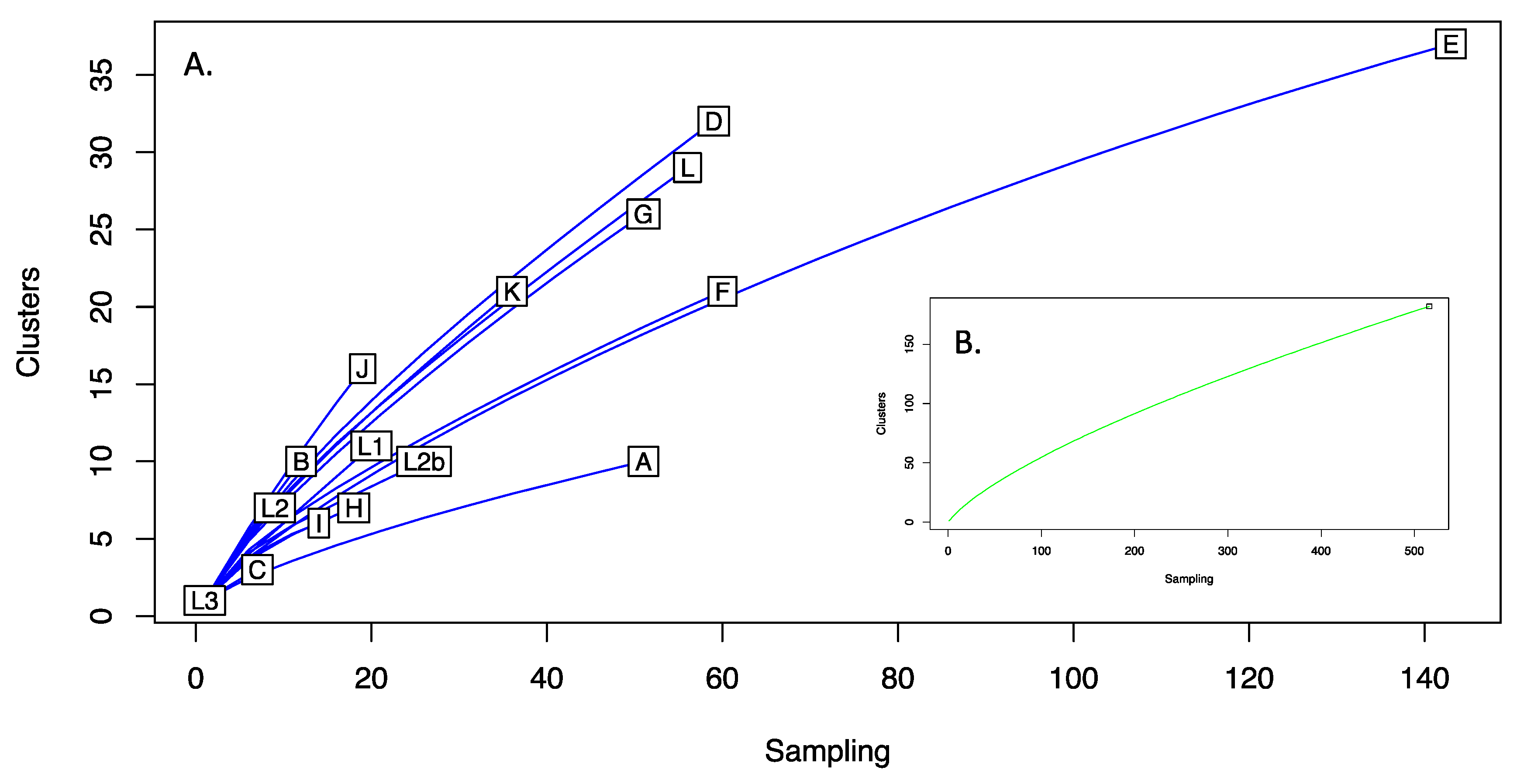
3.8. Clustering and Rarefaction Analysis of Plasmid Sequences
3.9. Number of 22 bp Repeats
4. Discussion
4.1. Nature and Extent of C. trachomatis Plasmid Diversity
4.2. Implications on Diagnostic Target Choice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Taylor, H.R. Trachoma. A Blinding Scourge from the Bronze Age to the Twenty First Century, 1st ed.; Haddington Press: Melbourne, Australia, 2008. [Google Scholar]
- Wong, W.F.; Chambers, J.P.; Gupta, R.; Arulanandam, B.P. Chlamydia and Its Many Ways of Escaping the Host Immune System. J. Pathog. 2019, 2019, 8604958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett, M.; Kuo, K.K.; Holmes, K.; Falkow, S. Plasmids of the genus Chlamydia. In Current Chemotherapy and Infectious Diseases; Nelson, J., Grassi, C., Eds.; American Society for Microbiology: Washington, DC, USA, 1980; Volume 2, pp. 1250–1252. [Google Scholar]
- Pickett, M.A.; Everson, J.S.; Pead, P.J.; Clarke, I.N. The plasmids of Chlamydia trachomatis and Chlamydophila pneumoniae (N16): Accurate determination of copy number and the paradoxical effect of plasmid-curing agents. Microbiology 2005, 151, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, S.; Cevenini, R.; Cosco, E.; Comanducci, M.; Ratti, G.; Scarlato, V. Transcriptional analysis of the Chlamydia trachomatis plasmid pCT identifies temporally regulated transcripts, anti-sense RNA and å 70 -selected promoters. Mol. Gen. Genet. 1993, 237, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Sharma, C.M.; Reinhardt, R.; Vogel, J.; Rudel, T. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 2010, 38, 868–877. [Google Scholar] [CrossRef]
- Ferreira, R.; Borges, V.; Nunes, A.; Borrego, M.J.; Gomes, J.P. Assessment of the load and transcriptional dynamics of Chlamydia trachomatis plasmid according to strains’ tissue tropism. Microbiol. Res. 2013, 168, 333–339. [Google Scholar] [CrossRef]
- Comanducci, M.; Cevenini, R.; Moroni, A.; Giuliani, M.M.; Ricci, S.; Scarlato, V.; Ratti, G. Expression of a plasmid gene of Chlamydia trachomatis encoding a novel 28 kDa antigen. J. Gen. Microbiol. 1993, 139, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Sriprakash, K.S.; MacAvoy, E.S. Characterization and sequence of a plasmid from the trachoma biovar of Chlamydia trachomatis. Plasmid 1987, 18, 205–214. [Google Scholar] [CrossRef]
- Hatt, C.; Ward, M.E.; Clarke, I.N. Analysis of the entire nucleotide sequence of the cryptic plasmid of Chlamydia trachomatis serovar L1. Evidence for involvement in DNA replication. Nucleic Acids Res. 1988, 16, 4053. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.E.; Davis, C.H.; Thresher, R.J.; Wyrick, P.B. Location of the origin of replication for the 7.5-kb Chlamydia trachomatis plasmid. Plasmid 1992, 27, 231–236. [Google Scholar] [CrossRef]
- Konieczny, I.; Bury, K.; Wawrzycka, A.; Wegrzyn, K. Iteron Plasmids. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novick, R.P. Contrasting lifestyles of rolling-circle phages and plasmids. Trends Biochem. Sci. 1998, 23, 434–438. [Google Scholar] [CrossRef]
- Park, K.; Han, E.; Paulsson, J.; Chattoraj, D.K. Origin pairing (‘handcuffing’) as a mode of negative control of P1 plasmid copy number. EMBO J. 2001, 20, 7323–7332. [Google Scholar] [CrossRef] [PubMed]
- Seth-Smith, H.M.B.; Harris, S.R.; Persson, K.; Marsh, P.; Barron, A.; Bignell, A.; Bjartling, C.; Clark, L.; Cutcliffe, L.T.; Lambden, P.R.; et al. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genom. 2009, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.S.; Lusher, M.; Storey, C.C.; Clarke, I.N. Plasmid diversity in Chlamydia. Microbiology 1997, 143, 1847–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filutowicz, M.; McEachern, M.J.; Helsinki, D.R. Positive and negative roles of an initiator protein at an origin of replication. Proc. Natl. Acad. Sci. USA 1986, 83, 9645–9649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Carlson, J.H.; Whitmire, W.M.; Kari, L.; Virtaneva, K.; Sturdevant, D.E.; Watkins, H.; Zhou, B.; Sturdevant, G.L.; Porcella, S.F.; et al. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect. Immun. 2013, 81, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cutcliffe, L.T.; Skilton, R.J.; Ramsey, K.H.; Thomson, N.R.; Clarke, I.N. The genetic basis of plasmid tropism between Chlamydia trachomatis and Chlamydia muridarum. Pathog. Dis. 2014, 72, 19–23. [Google Scholar] [CrossRef]
- Galaleldeen, A.; Taylor, A.B.; Chen, D.; Schuermann, J.P.; Holloway, S.P.; Hou, S.; Gong, S.; Zhong, G.; Hart, P.J. Structure of the Chlamydia trachomatis immunodominant antigen Pgp3. J. Biol. Chem. 2013, 288, 22068–22079. [Google Scholar] [CrossRef] [Green Version]
- Khurshid, S.; Govada, L.; Wills, G.; McClure, M.O.; Helliwell, J.R.; Chayen, N.E. Chlamydia protein Pgp3 studied at high resolution in a new crystal form. IUCrJ 2018, 5, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chen, D.; Zhong, Y.; Wang, S.; Zhong, G. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect. Immun. 2008, 76, 3415–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Lei, L.; Lu, C.; Galaleldeen, A.; Hart, P.J.; Zhong, G. Characterization of Pgp3, a Chlamydia trachomatis plasmid-encoded immunodominant antigen. J. Bacteriol. 2010, 192, 6017–6024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Huang, Y.; Yang, Z.; Sun, Y.; Gong, S.; Hou, S.; Chen, C.; Li, Z.; Liu, Q.; Wu, Y.; et al. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect. Immun. 2014, 82, 5327–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Sun, Y.; Qin, T.; Liu, Y. The Structural Integrity of Plasmid-Encoded Pgp3 Is Essential for Induction of Hydrosalpinx by Chlamydia muridarum. Front. Cell. Infect. Microbiol. 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.H.; Schripsema, J.H.; Smith, B.J.; Wang, Y.; Jham, B.C.; O’Hagan, K.P.; Thomson, N.R.; Murthy, A.K.; Skilton, R.J.; Chu, P.; et al. Plasmid CDS5 influences infectivity and virulence in a mouse model of Chlamydia trachomatis urogenital infection. Infect. Immun. 2014, 82, 3341–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, P.J.; Wills, G.S.; Righarts, A.; Vieira, S.; Kounali, D.; Samuel, D.; Winston, A.; Muir, D.; Dickson, N.P.; McClure, M.O. Chlamydia trachomatis Pgp3 Antibody Persists and Correlates with Self-Reported Infection and Behavioural Risks in a Blinded Cohort Study. PLoS ONE 2016, 11, e0151497. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Dong, X.; Yang, Z.; Li, Z.; Liu, Q.; Zhong, G. Chlamydial plasmid-encoded virulence factor Pgp3 neutralizes the antichlamydial activity of human cathelicidin LL-37. Infect. Immun. 2015, 83, 4701–4709. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Zhang, T.; Melero, J.; Huang, Y.; Liu, Y.; Liu, Q.; He, C.; Nelson, D.E.; Zhong, G. The Genital Tract Virulence Factor pGP3 Is Essential for Chlamydia muridarum Colonization in the Gastrointestinal Tract. Infect. Immun. 2018, 86, e00429-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Huo, Z.; Ma, J.; He, C.; Zhong, G. The Plasmid-Encoded pGP3 Promotes Chlamydia Evasion of Acidic Barriers in Both Stomach and Vagina. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Lei, W.; Su, S.; Bu, J.; Zhu, S.; Huang, Q.; Li, Z. Chlamydia trachomatis plasmid-encoded protein Pgp3 inhibits apoptosis via the PI3K-AKT-mediated MDM2-p53 axis. Mol. Cell. Biochem. 2019, 452, 167–176. [Google Scholar] [CrossRef]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Clarke, I.N. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011, 7, e1002258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, C.; Gong, S.; Hou, S.; Qi, M.; Liu, Q.; Baseman, J.; Zhong, G. Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J. Bacteriol. 2014, 196, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; Clarke, I.N.; Seth-Smith, H.M.; Solomon, A.W.; Cutcliffe, L.T.; Marsh, P.; Skilton, R.J.; Holland, M.J.; Mabey, D.; Peeling, R.W.; et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 2012, 44, 413–419, S411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.R.; Thomas, C.M. Active partitioning of bacterial plasmids. J. Gen. Microbiol. 1992, 138, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, E.M.; Markoff, B.A.; Schachter, J.; de la Maza, L.M. The 7.5-kb plasmid present in Chlamydia trachomatis is not essential for the growth of this microorganism. Plasmid 1990, 23, 144–148. [Google Scholar] [CrossRef]
- Carlson, J.H.; Whitmire, W.M.; Crane, D.D.; Wicke, L.; Virtaneva, K.; Sturdevant, D.E.; Kupko, J.J., III; Porcella, S.F.; Martinez-Orengo, N.; Heinzen, R.A.; et al. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect. Immun. 2008, 76, 2273–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares-Zavaleta, N.; Whitmire, W.; Gardner, D.; Caldwell, H.D. Immunization with the attenuated plasmidless Chlamydia trachomatis L2(25667R) strain provides partial protection in a murine model of female genitourinary tract infection. Vaccine 2010, 28, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Sigar, I.M.; Schripsema, J.H.; Wang, Y.; Clarke, I.N.; Cutcliffe, L.T.; Seth-Smith, H.M.B.; Thomson, N.R.; Bjartling, C.; Unemo, M.; Persson, K.; et al. Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathog. Dis. 2014, 70, 61–69. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, C.M.; Ingalls, R.R.; Andrews, C.W.; Scurlock, A.M.; Darville, T. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J. Immunol. 2007, 179, 4027–4034. [Google Scholar] [CrossRef] [Green Version]
- Kari, L.; Whitmire, W.M.; Olivares-Zavaleta, N.; Goheen, M.M.; Taylor, L.D.; Carlson, J.H.; Sturdevant, G.L.; Lu, C.; Bakios, L.E.; Randall, L.B.; et al. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J. Exp. Med. 2011, 208, 2217–2223. [Google Scholar] [CrossRef] [Green Version]
- Farencena, A.; Comanducci, M.; Donati, M.; Ratti, G.; Cevenini, R. Characterization of a new isolate of Chlamydia trachomatis which lacks the common plasmid and has properties of Biovar trachoma. Infect. Immun. 1997, 65, 2965–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, D.R.; Williams, J.A.; Van Der Pol, B.; Jones, R.B. Identification of a Chlamydia trachomatis serovar E urogenital isolate which lacks the cryptic plasmid. Infect. Immun 1998, 66, 6010–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Persson, K.; Bjartling, C.; Clarke, I.N. Genetic transformation of a clinical (genital tract), plasmid-free isolate of Chlamydia trachomatis: Engineering the plasmid as a cloning vector. PLoS ONE 2013, 8, e59195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cutcliffe, L.T.; Skilton, R.J.; Persson, K.; Bjartling, C.; Clarke, I.N. Transformation of a plasmid-free, genital tract isolate of Chlamydia trachomatis with a plasmid vector carrying a deletion in CDS6 revealed that this gene regulates inclusion phenotype. Pathog. Dis. 2013, 67, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.E.; Wolf, K.; Fields, K.A. Gene Deletion by Fluorescence-Reported Allelic Exchange Mutagenesis in Chlamydia trachomatis. mBio 2016, 7, e01817-15. [Google Scholar] [CrossRef] [Green Version]
- Ripa, T.; Nilsson, P.A. A Chlamydia trachomatis strain with a 377-bp deletion in the cryptic plasmid causing false-negative nucleic acid amplification tests. Sex. Transm. Dis. 2007, 34, 255–256. [Google Scholar] [CrossRef]
- Hadfield, J.; Harris, S.R.; Seth-Smith, H.M.B.; Parmar, S.; Andersson, P.; Giffard, P.M.; Schachter, J.; Moncada, J.; Ellison, L.; Vaulet, M.L.G.; et al. Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Skilton, R.J.; Cutcliffe, L.T.; Pickett, M.A.; Lambden, P.R.; Fane, B.A.; Clarke, I.N. Intracellular parasitism of chlamydiae: Specific infectivity of chlamydiaphage Chp2 in Chlamydophila abortus. J. Bacteriol. 2007, 189, 4957–4959. [Google Scholar] [CrossRef] [Green Version]
- Seth-Smith, H.M.B.; Harris, S.R.; Scott, P.; Parmar, S.; Marsh, P.; Unemo, M.; Clarke, I.N.; Parkhill, J.; Thomson, N.R. Generating whole bacterial genome sequences of low-abundance species from complex samples with IMS-MDA. Nat. Protoc. 2013, 8, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.T.; Brown, A.C.; Kundu, S.; Tutill, H.J.; Williams, R.; Brown, J.R.; Holdstock, J.; Holland, M.J.; Stevenson, S.; Dave, J.; et al. Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect. Dis. 2014, 14, 591. [Google Scholar] [CrossRef]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Evolutionary trees from gene frequencies and quantitative characters: Finding maximum likelihood estimates. Evolution 1981, 35, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Gojobori, T. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 1999, 16, 1315–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Comanducci, M.; Ricci, S.; Cevenini, R.; Ratti, G. Diversity of the Chlamydia trachomatis common plasmid in biovars with different pathogenicity. Plasmid 1990, 23, 149–154. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. CD-HIT: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. R package version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 17 February 2020).
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Versteeg, B.; Bruisten, S.M.; Pannekoek, Y.; Jolley, K.A.; Maiden, M.C.J.; van der Ende, A.; Harrison, O.B. Genomic analyses of the Chlamydia trachomatis core genome show an association between chromosomal genome, plasmid type and disease. BMC Genom. 2018, 19, 130. [Google Scholar] [CrossRef] [Green Version]
- Borges, V.; Gomes, J.P. Deep comparative genomics among Chlamydia trachomatis lymphogranuloma venereum isolates highlights genes potentially involved in pathoadaptation. Infect. Genet. Evol. 2015, 32, 74–88. [Google Scholar] [CrossRef]
- Rockey, D.D. Unraveling the basic biology and clinical significance of the chlamydial plasmid. J. Exp. Med. 2011, 208, 2159–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alff-Steinberger, C. The genetic code and error transmission. Proc. Natl. Acad. Sci. USA 1969, 64, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Aiba, H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 2007, 10, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Wassarman, K.M.; Storz, G. 6S RNA regulates E. coli RNA polymerase activity. Cell 2000, 101, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Oogai, Y.; Gotoh, Y.; Ogura, Y.; Kawada-Matsuo, M.; Hayashi, T.; Komatsuzawa, H. Small RNA repertoires and their intraspecies variation in Aggregatibacter actinomycetemcomitans. DNA Res. 2018, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.I.; Dooley, C.A.; Fischer, E.R.; Scidmore, M.A.; Fields, K.A.; Hackstadt, T. Three temporal classes of gene expression during the Chlamydia trachomatis developmental cycle. Mol. Microbiol. 2000, 37, 913–925. [Google Scholar] [CrossRef]
- Nicholson, T.L.; Olinger, L.; Chong, K.; Schoolnik, G.; Stephens, R.S. Global Stage-Specific Gene Regulation during the Developmental Cycle of Chlamydia trachomatis. J. Bacteriol. 2003, 185, 3179–3189. [Google Scholar] [CrossRef] [Green Version]
- Belland, R.J.; Nelson, D.E.; Virok, D.; Crane, D.D.; Hogan, D.; Sturdevant, D.; Beatty, W.L.; Caldwell, H.D. Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc. Natl. Acad. Sci. USA 2003, 100, 15971–15976. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.; Borrego, M.J.; Gomes, J.P. Genomic features beyond Chlamydia trachomatis phenotypes: What do we think we know? Infect. Genet. Evol. 2013, 16, 392–400. [Google Scholar] [CrossRef]
- Abdelsamed, H.; Peters, J.; Byrne, G.I. Genetic variation in Chlamydia trachomatis and their hosts: Impact on disease severity and tissue tropism. Future Microbiol. 2013, 8, 1129–1146. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Antelo, M.; Nunes, A.; Borges, V.; Damiao, V.; Borrego, M.J.; Gomes, J.P. In silico scrutiny of genes revealing phylogenetic congruence with clinical prevalence or tropism properties of Chlamydia trachomatis strains. G3 (Bethesda) 2014, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, P.; Harris, S.R.; Seth Smith, H.M.; Hadfield, J.; O’Neill, C.; Cutcliffe, L.T.; Douglas, F.P.; Asche, L.V.; Mathews, J.D.; Hutton, S.I.; et al. Chlamydia trachomatis from Australian Aboriginal people with trachoma are polyphyletic composed of multiple distinctive lineages. Nat. Commun. 2016, 7, 10688. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Ripa, T.; Nilsson, P. A variant of Chlamydia trachomatis with deletion in cryptic plasmid: Implications for use of PCR diagnostic tests. Euro. Surveill. 2006, 11, E061109. [Google Scholar] [CrossRef] [PubMed]
- Hokynar, K.; Rantakokko-Jalava, K.; Hakanen, A.; Havana, M.; Mannonen, L.; Jokela, P.; Kurkela, S.; Lappalainen, M.; Unemo, M.; Puolakkainen, M. The Finnish New Variant of Chlamydia trachomatis with a Single Nucleotide Polymorphism in the 23S rRNA Target Escapes Detection by the Aptima Combo 2 Test. Microorganisms 2019, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Unemo, M.; Hansen, M.; Hadad, R.; Lindroth, Y.; Fredlund, H.; Puolakkainen, M.; Sundqvist, M. Finnish new variant of Chlamydia trachomatis escaping detection in the Aptima Combo 2 assay also present in Orebro County, Sweden, May 2019. Euro Surveill. 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- Yeow, T.C.; Wong, W.F.; Sabet, N.S.; Sulaiman, S.; Shahhosseini, F.; Tan, G.M.; Movahed, E.; Looi, C.Y.; Shankar, E.M.; Gupta, R.; et al. Prevalence of plasmid-bearing and plasmid-free Chlamydia trachomatis infection among women who visited obstetrics and gynecology clinics in Malaysia. BMC Microbiol. 2016, 16, 45. [Google Scholar] [CrossRef] [Green Version]
- Magbanua, J.P.; Goh, B.T.; Michel, C.E.; Aguirre-Andreasen, A.; Alexander, S.; Ushiro-Lumb, I.; Ison, C.; Lee, H. Chlamydia trachomatis variant not detected by plasmid based nucleic acid amplification tests: Molecular characterisation and failure of single dose azithromycin. Sex. Transm. Infect. 2007, 83, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, K.; Prakash, P.; Rani, A.; Singh, S.K. Multiplex nested polymerase chain reaction targeting multiple genes for the detection of Neisseria gonorrhoeae and Chlamydia trachomatis in genitourinary specimens. Indian J. Sex. Transm. Dis. Aids 2019, 40, 152–158. [Google Scholar] [CrossRef]
- Ma, C.; Du, J.; He, W.; Chen, R.; Li, Y.; Dou, Y.; Yuan, X.; Zhao, L.; Gong, H.; Liu, P.; et al. Rapid and accurate diagnosis of Chlamydia trachomatis in the urogenital tract by a dual-gene multiplex qPCR method. J. Med. Microbiol. 2019, 68, 1732–1739. [Google Scholar] [CrossRef]
- Frej-Madrzak, M.; Grybos, A.; Grybos, M.; Teryks-Wolyniec, D.; Jama-Kmiecik, A.; Sarowska, J.; Choroszy-Krol, I. PCR diagnostics of Chlamydia trachomatis in asymptomatic infection by women. Ginekol. Pol. 2018, 89, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, M.J.; Chen, C.Y.; Yang, C.; McCorrister, S.; Grant, C.; Westmacott, G.; Yuan, X.Y.; Ochoa, E.; Fariss, R.; Whitmire, W.M.; et al. Plasmid Negative Regulation of CPAF Expression Is Pgp4 Independent and Restricted to Invasive Chlamydia trachomatis Biovars. mBio 2018, 9, e02164-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, B.; Torner, A.; Low, N.; Klint, M.; Nilsson, A.; Velicko, I.; Soderblom, T.; Blaxhult, A. Emergence and spread of Chlamydia trachomatis variant, Sweden. Emerg. Infect. Dis. 2008, 14, 1462–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workowski, K.A.; Bolan, G.A. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm. Rep. 2015, 64, 1–137. [Google Scholar]
- McLean, C.A.; Stoner, B.P.; Workowski, K.A. Treatment of lymphogranuloma venereum. Clin. Infect. Dis. 2007, 44 (Suppl. 3), S147–S152. [Google Scholar] [CrossRef] [Green Version]
- Twin, J.; Stevens, M.P.; Garland, S.M.; Zaia, A.M.; Tabrizi, S.N. Rapid determination of lymphogranuloma venereum serovars of Chlamydia trachomatis by quantitative high-resolution melt analysis (HRMA). J. Clin. Microbiol. 2012, 50, 3751–3753. [Google Scholar] [CrossRef] [Green Version]
- Quint, K.D.; Bom, R.J.; Bruisten, S.M.; van Doorn, L.J.; Nassir Hajipour, N.; Melchers, W.J.; de Vries, H.J.; Morre, S.A.; Quint, W.G. Comparison of three genotyping methods to identify Chlamydia trachomatis genotypes in positive men and women. Mol. Cell. Probes 2010, 24, 266–270. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| CDS (Pgp) | Function of Encoded Protein | Summary of Current Knowledge and References |
|---|---|---|
| 1 (7) | Plasmid Replication | Homologue of integrase, part of the family of phage proteins [17]. Role in regulation of plasmid replication [18]. Loss of function does not affect plasmid maintenance due to functional redundancy with CDS2 [16,17,19]. |
| 2 (8) | Plasmid Replication | Homologue of recombinase, part of the family of phage proteins; role in regulation of plasmid replication [17]. Critical for plasmid maintenance [19]. The main determinant of plasmid tropism [20]. |
| 3 (1) | Plasmid Replication | Homology observed with DnaB helicase proteins of Escherichia coli and Salmonella typhimurium, helicase involved in unwinding of double stranded DNA [11,17]. Critical for plasmid maintenance [19]. |
| 4 (2) | Function unknown | Required for plasmid maintenance [19]. |
| 5 (3) | Virulence protein | 28 kDa protein [9]. CDS3-encoded Pgp (plasmid glycoprotein) 3 crystal structure resolved for genotypes D and L2 [21,22]. Dispensable for chlamydial growth in vitro [19]. Shown to be secreted from the chlamydial inclusion into the cytosol of the host cell [23,24]. Pgp3 from C. muridarum shown as major virulence factor responsible for hydrosalpynx induction in mice [25,26]. Strong immunogenic properties; purified protein stimulated macrophages to release inflammatory cytokines in the mouse model and acts as Toll-like receptor 4 (TLR4), suggesting a role in Chlamydia-induced inflammatory pathology [23]. Later shown to have an essential role in virulence and infectivity in vivo [27]. Pgp3 antibody was found to persist for at least 12 years post infection, suggesting a role for C. trachomatis Pgp3 serology in evaluating control programmes [28]. C. trachomatis Pgp3 neutralizes the antichlamydial activity of human cathelicidin LL-37 [29] and is essential for colonisation of the gastrointestinal tract [30] due to evasion of acidic barriers (in both stomach and vagina) [31]. Pgp3 expression is also shown to inhibit apoptosis via the PI3K-AKT-mediated MDM2-p53 axis [32]. |
| 6 (4) | Transcriptional regulation | Role in ability of C. trachomatis to accumulate glycogen [33]. Transcriptional regulation of the plasmid virulence protein Pgp3 and of chromosomal gene expression [19,25]. |
| 7 (5) | Regulation of partitioning and copy number | Partial homology to E. coli plasmid and phage encoded proteins, including SopA and ParA, which are involved in partitioning and copy number in E. coli [17]. Shown to negatively regulate some plasmid-dependant genes in C. muridarum [34]. Dispensable for chlamydial growth in cell culture [19]. |
| 8 (6) | Regulation of partitioning and copy number | Thought to function in conjunction with pCDS7 in a similar manner to that of the sopA/B and parA/B operons in E. coli [17]. The only CDS which has a homologue on the C. trachomatis chromosome, which is also present in plasmid free isolates [19]. Critical for plasmid maintenance [19]. |
| Length (bp) | Number of Intragenic SNP Loci | SNP loci Rate (%) | Total SNPs | Average Number of SNPs per Locus | Non Synonymous (NS) SNPs (%) | NS SNPs Involving a Change of Amino Acid Characteristics (%) | |
|---|---|---|---|---|---|---|---|
| CDS | |||||||
| 1 | 918 | 30 | 3.27 | 2012 | 67.06 | 762 (37.9) | 436 (57.2) |
| 2 | 993 | 26 | 2.62 | 1734 | 66.69 | 364 (21) | 237 (65.1) |
| 3 | 1356 | 32 | 2.36 | 2116 | 66.13 | 575 (27.2) | 182 (31.7) |
| 4 | 1065 | 27 | 2.54 | 1435 | 53.14 | 518 (36.1) | 339 (65.4) |
| 5 | 795 | 28 | 3.52 | 2267 | 80.96 | 1335 (58.9) | 850 (63.4) |
| 6 | 309 | 6 | 1.94 | 85 | 14.16 | 55 (64.7) | 53 (96.4) |
| 7 | 825 | 21 | 2.55 | 1072 | 51.05 | 928 (86.6) | 722 (77.8) |
| 8 | 744 | 29 | 3.90 | 1126 | 38.83 | 450 (40) | 252 (56) |
| Total | 7005 | 199 | 2.84 | 11,847 | - | 4987 (42.1) | 3070 (61.6) |
| Nucleotide Sequences | Amino Acid Sequences | |||
|---|---|---|---|---|
| CDS | d | SE | d | SE |
| 1 | 0.004 | 0.001 | 0.003 | 0.002 |
| 2 | 0.004 | 0.001 | 0.001 | 0.001 |
| 3 | 0.003 | 0.001 | 0.003 | 0.001 |
| 4 | 0.004 | 0.001 | 0.003 | 0.002 |
| 5 | 0.007 | 0.002 | 0.013 | 0.004 |
| 6 | 0.001 | 0.001 | 0.002 | 0.002 |
| 7 | 0.003 | 0.001 | 0.008 | 0.003 |
| 8 | 0.004 | 0.001 | 0.005 | 0.002 |
| Plasmid | 0.004 | 0.000 | N/A | N/A |
| Number of SNP Loci (%) | Total Number of SNPs (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| CDS | Base 1 | Base 2 | Base 3 | Total | Base 1 | Base 2 | Base 3 | Total |
| 1 | 16 (53.3) | 4 (13.3) | 10 (33.3) | 30 | 470 (23.4) | 292 (14.5) | 1250 (62.1) | 2012 |
| 2 | 4 (15.4) | 4 (15.4) | 18 (69.2) | 26 | 129 (7.4) | 39 (2.2) | 1566 (90.3) | 1734 |
| 3 | 10 (31.25) | 6 (18.75) | 16 (50) | 32 | 164 (7.8) | 408 (19.3) | 1544 (73) | 2116 |
| 4 | 6 (22.2) | 1 (3.7) | 20 (70.1) | 27 | 426 (29.7) | 1 (0.1) | 1008 (70.2) | 1435 |
| 5 | 9 (32.1) | 8 (28.6) | 11 (39.3) | 28 | 594 (26.2) | 633 (27.9) | 1040 (45.9) | 2267 |
| 6 | 2 (33.3) | 1 (16.7) | 3 (50) | 6 | 2 (2.4) | 1 (1.2) | 82 (96.5) | 85 |
| 7 | 5 (23.8) | 7 (33.3) | 9 (42.8) | 21 | 300 (28) | 626 (58.4) | 146 (13.6) | 1072 |
| 8 | 4 (13.8) | 8 (27.6) | 17 (58.6) | 29 | 114 (10.1) | 196 (17.4) | 816 (72.5) | 1126 |
| TOTAL | 56 (28.1) | 39 (19.6) | 104 (52.3) | 199 | 2199 (18.6) | 2196 (18.5) | 7452 (62.9) | 11,847 |
| Open Reading Frame | Base | Number of Sequences with SNP | Reference Code | SNP Change | Pre-AA | Post-AA | Type of Stop Codon (Premature or Delayed) | Change to Size of CDS |
|---|---|---|---|---|---|---|---|---|
| 1 | 1080 | 7 | TGA | GGA | STOP | G | Delayed | +3 codons |
| 4 | 4667 | 33 | GAA | TAA | E | STOP | Premature | −4 codons |
| 4 | 4679 | 2 | TAA | CAA | STOP | Q | Delayed | +1 codon |
| Number of Repeats | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Geno-Type | Four Repeats | Three + imp. | Three Repeats | Two + imp. | Two Repeats | One + imp. | One Repeat | None + imp. | No Repeats | Total |
| A | 0 | 45 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 47 |
| B | 13 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14 |
| Ba | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| C | 5 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
| D | 32 | 11 | 5 | 1 | 1 | 6 | 0 | 0 | 0 | 56 |
| E | 66 | 58 | 0 | 1 | 0 | 16 | 0 | 0 | 2 | 143 |
| F | 27 | 20 | 3 | 1 | 0 | 9 | 0 | 0 | 0 | 60 |
| G | 33 | 10 | 5 | 2 | 1 | 4 | 0 | 0 | 0 | 55 |
| H | 16 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 18 |
| I | 7 | 3 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 12 |
| Ia | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 |
| J | 8 | 7 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 18 |
| K | 19 | 8 | 2 | 2 | 2 | 3 | 0 | 0 | 0 | 36 |
| L1 | 12 | 5 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 20 |
| L2 | 7 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 |
| L2b | 12 | 2 | 0 | 1 | 0 | 9 | 0 | 1 | 0 | 25 |
| L3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Total | 257 | 177 | 16 | 8 | 5 | 57 | 0 | 2 | 2 | 524 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, C.A.; Hadfield, J.; Thomson, N.R.; Cleary, D.W.; Marsh, P.; Clarke, I.N.; O’Neill, C.E. The Nature and Extent of Plasmid Variation in Chlamydia trachomatis. Microorganisms 2020, 8, 373. https://doi.org/10.3390/microorganisms8030373
Jones CA, Hadfield J, Thomson NR, Cleary DW, Marsh P, Clarke IN, O’Neill CE. The Nature and Extent of Plasmid Variation in Chlamydia trachomatis. Microorganisms. 2020; 8(3):373. https://doi.org/10.3390/microorganisms8030373
Chicago/Turabian StyleJones, Charlotte A., James Hadfield, Nicholas R. Thomson, David W. Cleary, Peter Marsh, Ian N. Clarke, and Colette E. O’Neill. 2020. "The Nature and Extent of Plasmid Variation in Chlamydia trachomatis" Microorganisms 8, no. 3: 373. https://doi.org/10.3390/microorganisms8030373
APA StyleJones, C. A., Hadfield, J., Thomson, N. R., Cleary, D. W., Marsh, P., Clarke, I. N., & O’Neill, C. E. (2020). The Nature and Extent of Plasmid Variation in Chlamydia trachomatis. Microorganisms, 8(3), 373. https://doi.org/10.3390/microorganisms8030373