The Interplay between Mucosal Microbiota Composition and Host Gene-Expression is Linked with Infliximab Response in Inflammatory Bowel Diseases

 ,
, 
 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
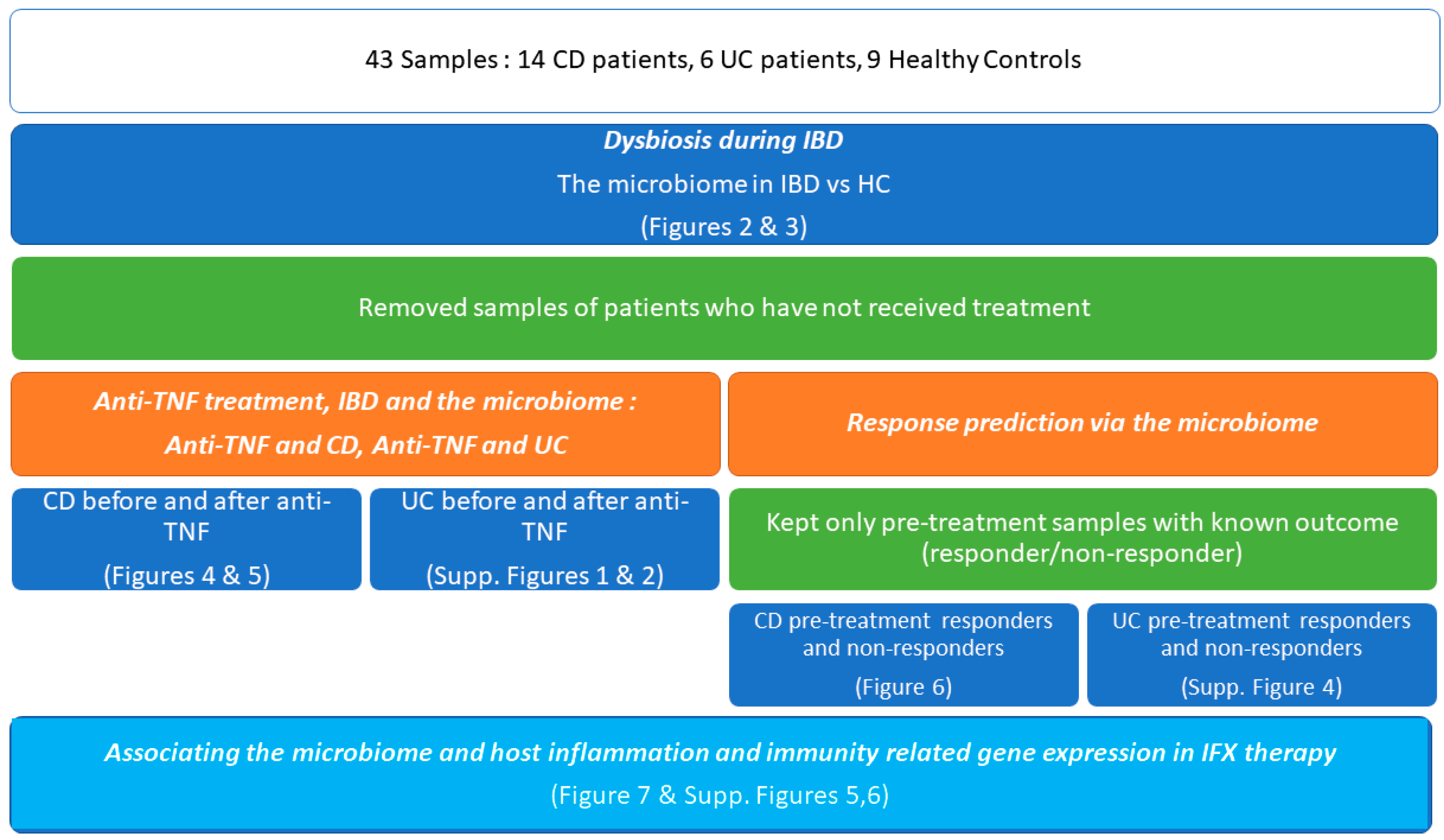
2. Materials and Methods
2.1.Samples
2.2. RNA Extraction and Gene Expression
2.3. Differential Gene Expression Analysis
- (1)
- Group 1: CD patients who responded to IFX treatment (5 samples at baseline)
- (2)
- Group 2: CD patients who responded to IFX treatment (5 samples after treatment)
- (3)
- Group 3: CD patients who did not respond to IFX treatment (5 samples at baseline)
- (4)
- Group 4: CD patients who did not respond to IFX treatment (5 samples after treatment)
- (5)
- Group 5: UC patients who responded to IFX treatment (2 samples at baseline)
- (6)
- Group 6: UC patients who responded to IFX treatment (2 samples after treatment)
- (7)
- Group 7: UC patients who did not respond to IFX treatment (2 samples at baseline)
- (8)
- Group 8: UC patients who did not respond to IFX treatment (2 samples after treatment)
2.4. DNA Extraction and 16S rRNA Amplicon Sequencing
2.5. Pre-Processing and OTU Picking
2.6. Downstream Analysis
3. Results
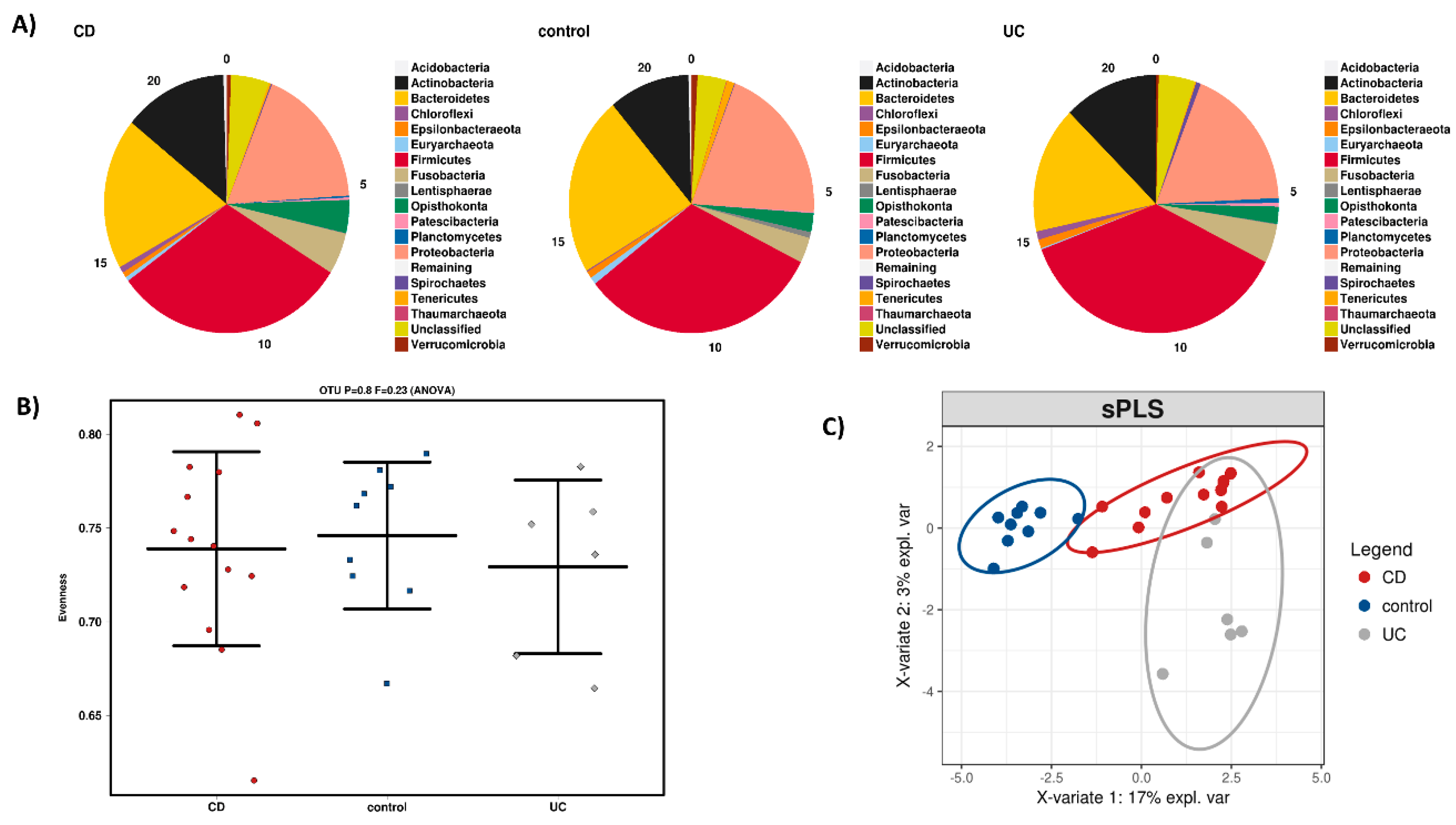
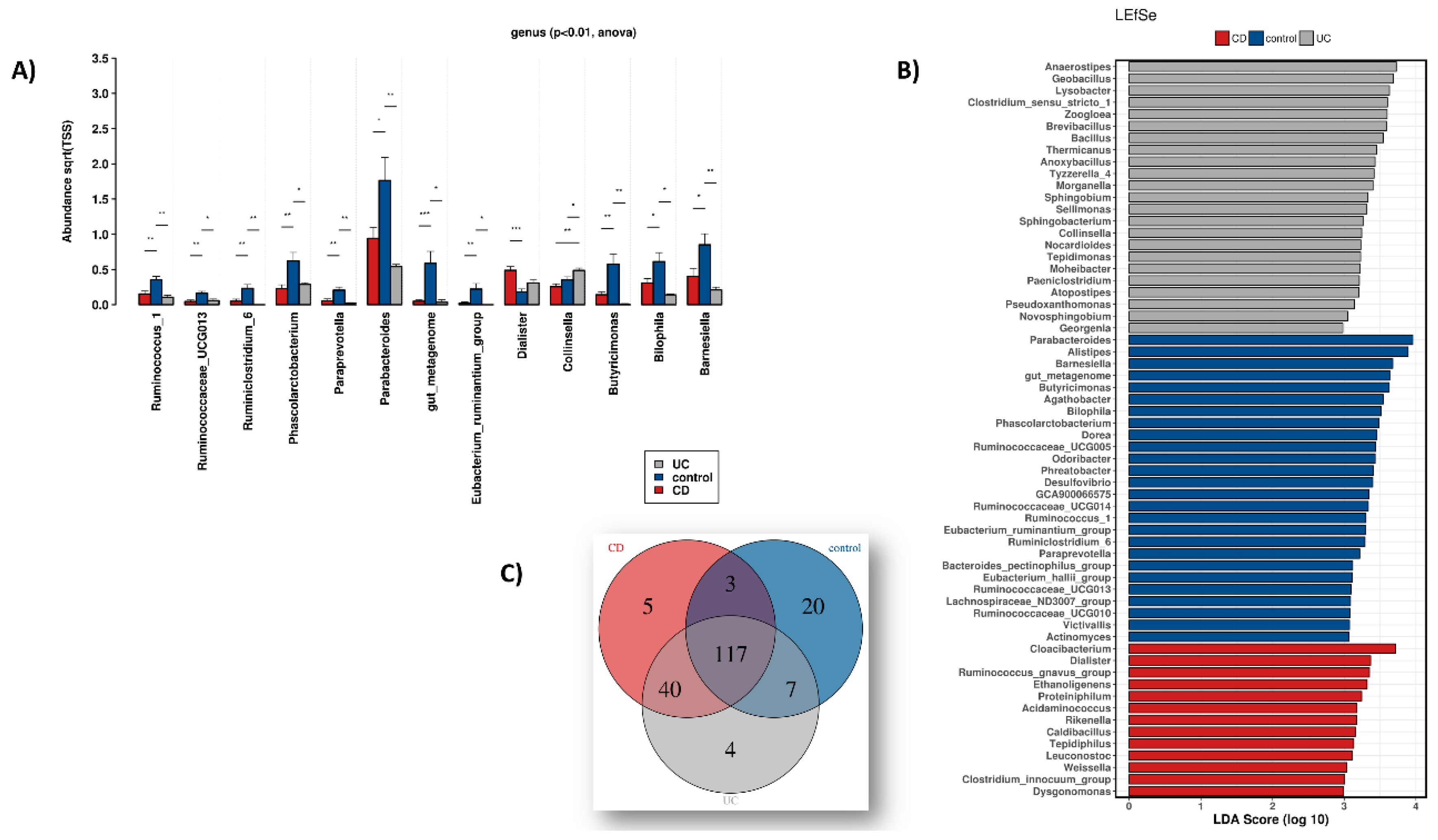
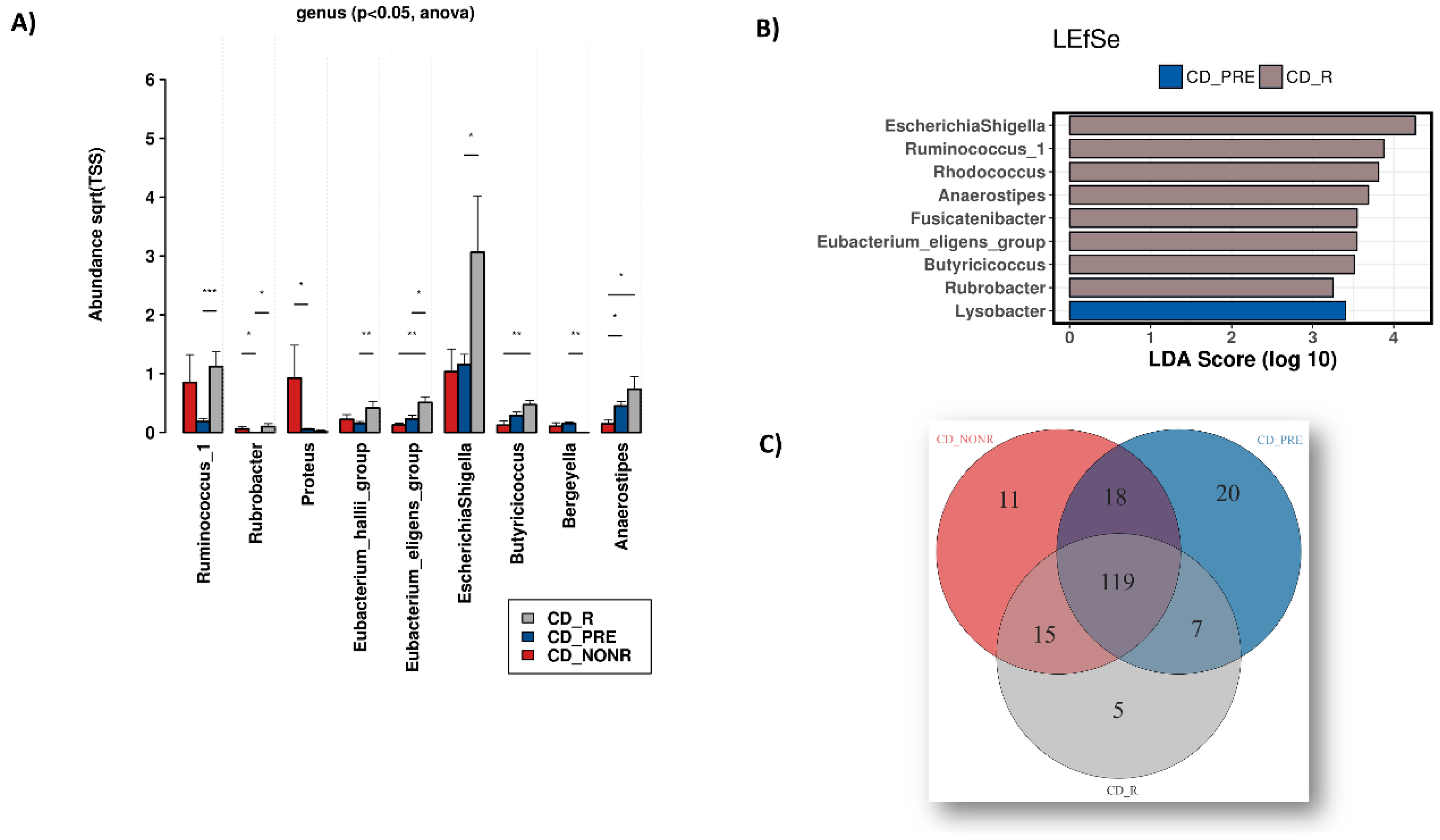
3.1. Dysbiosis during IBD
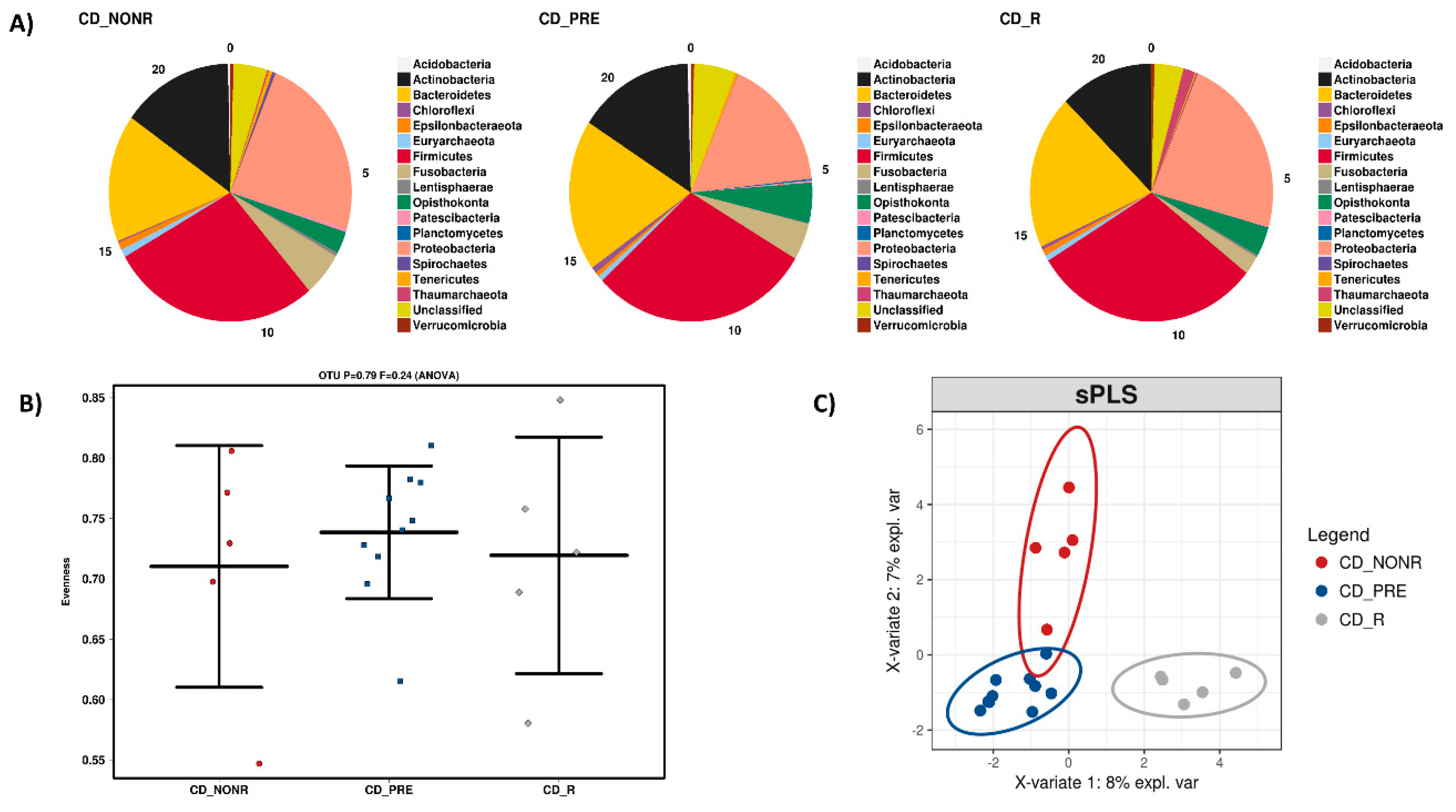
3.2. Anti-TNF Treatment, IBD and the Microbiome
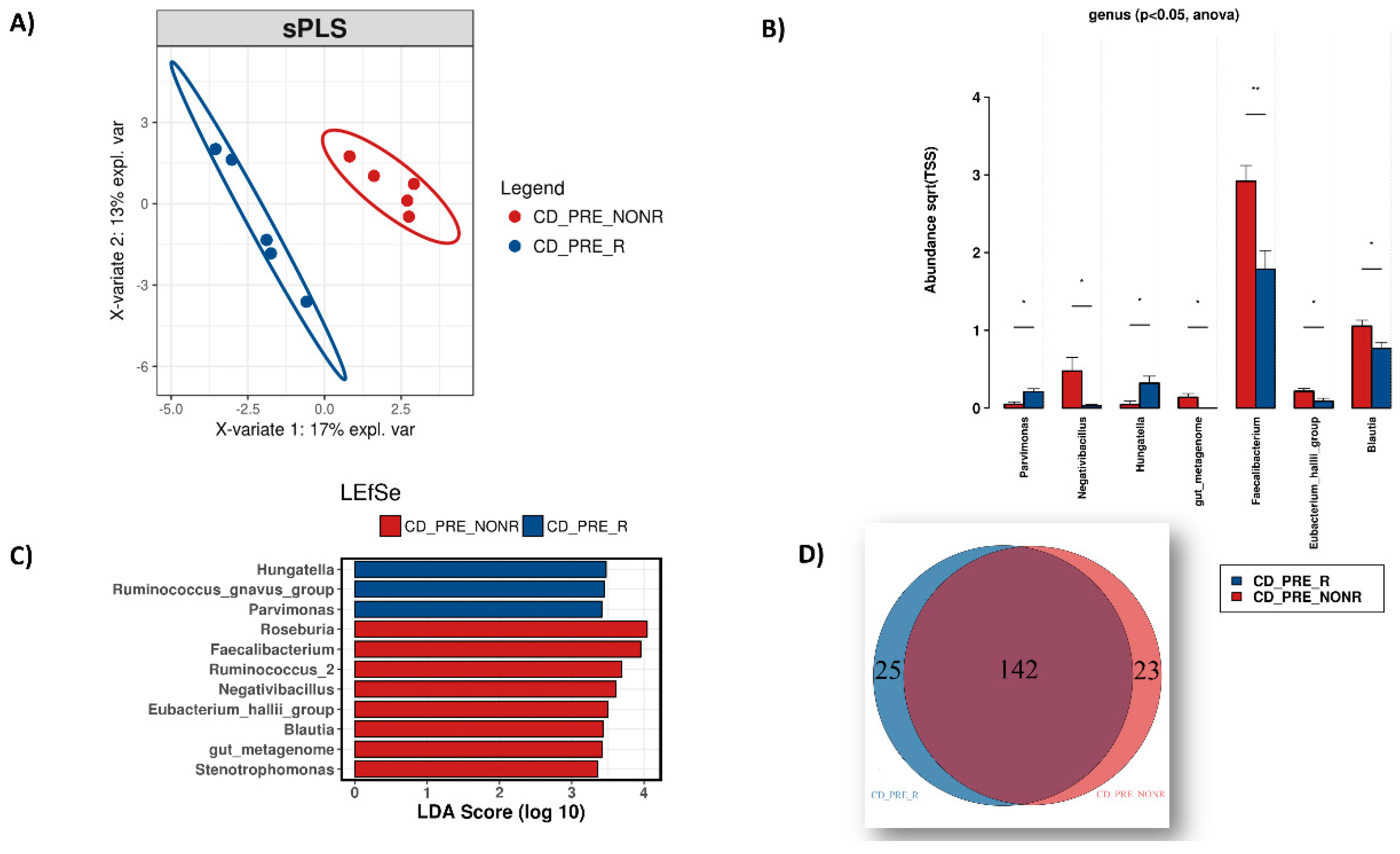
3.3. IFX Response Prediction via the Microbiome
3.4. Differential Gene Expression Analysis (DGEA) of Mucosal Tissue
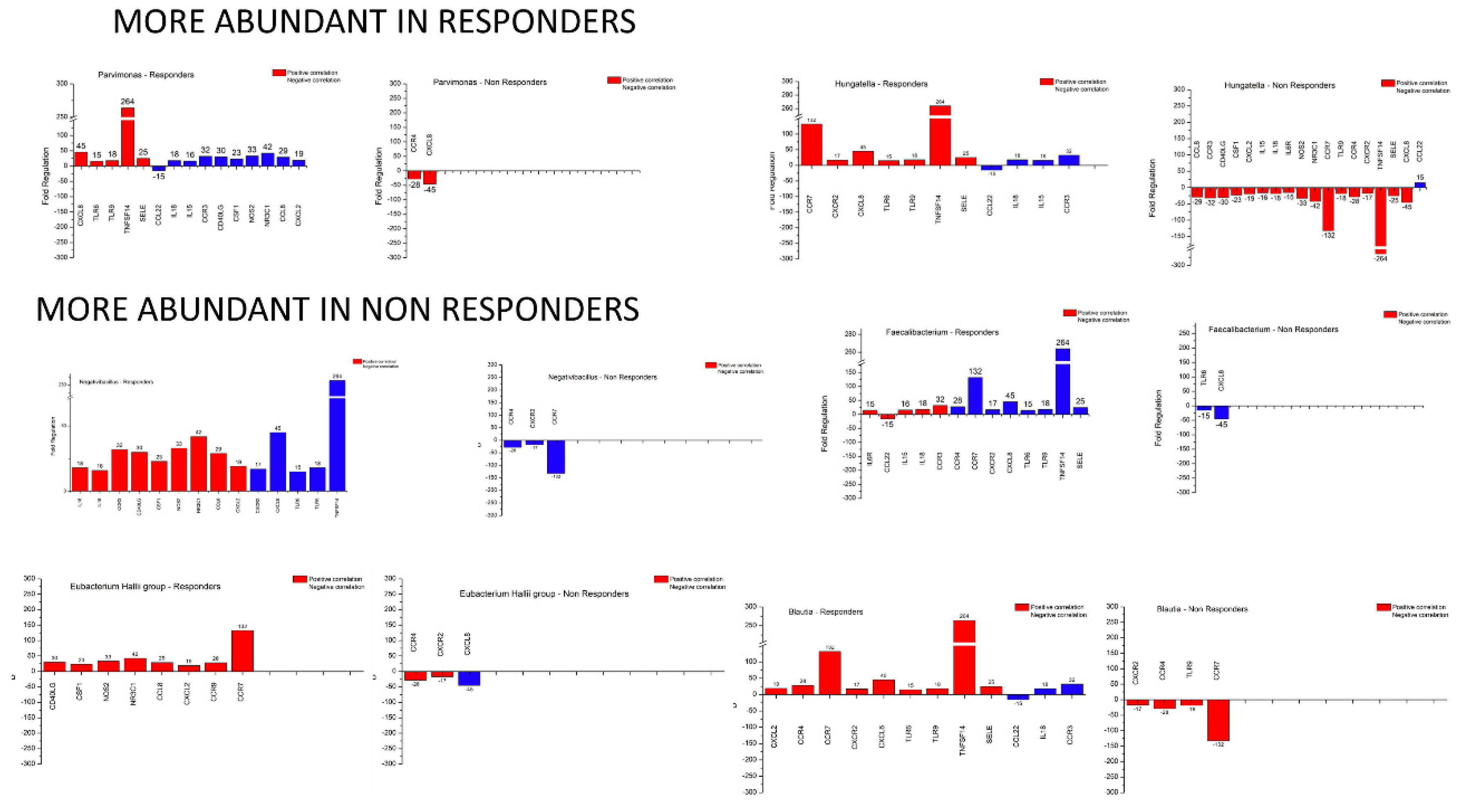
3.5. Microbiome – Host Gene Expression Associations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Legaki, E.; Gazouli, M. Influence of environmental factors in the development of inflammatory bowel diseases. WJGPT 2016, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Gazouli, M. Inflammatory bowel disease: Recent advances on genetics and innate immunity. Ann. Gastroenterol. 2011, 24, 164. [Google Scholar] [PubMed]
- Panagi, M.; Georgila, K.; Eliopoulos, A.G.; Apidianakis, Y. Constructing personalized longitudinal holo’omes of colon cancer-prone humans and their modeling in flies and mice. Oncotarget 2015, 10, 4224–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.J.; Cho, J.H.; Gevers, D.; Chu, H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 2174–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkouskou, K.; Deligianni, C.; Tsatsanis, C.; Eliopoulos, A.G. The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol. 2014, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, R.B. The intestinal microbiota in inflammatory bowel diseases. In Nutrition, Gut Microbiota and Immunity: Therapeutic Targets for IBD; Karger Publishers: Basel, Switerland, 2014; Volume 79, pp. 29–39. [Google Scholar]
- Vrakas, S.; Mountzouris, K.C.; Michalopoulos, G.; Karamanolis, G.; Papatheodoridis, G.; Tzathas, C.; Gazouli, M. Intestinal bacteria composition and translocation of bacteria in inflammatory bowel disease. PLoS ONE 2017, 12, e0170034. [Google Scholar] [CrossRef] [Green Version]
- Dovrolis, N.; Drygiannakis, I.; Filidou, E.; Kandilogiannakis, L.; Arvanitidis, K.; Tentes, I.; Kolios, G.; Valatas, V. Gut microbial signatures underline complicated crohn’s disease but vary between cohorts; An in silico approach. Inflamm. Bowel Dis. 2019, 25, 217–225. [Google Scholar] [CrossRef]
- Strober, W. Impact of the gut microbiome on mucosal inflammation. Trends Immunol. 2013, 34, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Papamichael, K.; Lin, S.; Moore, M.; Papaioannou, G.; Sattler, L.; Cheifetz, A.S. Infliximab in inflammatory bowel disease. Ther. Adv. Chronic Dis. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Chowers, Y. Tailoring anti-TNF therapy in IBD: Drug levels and disease activity. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, H.; Amital, H. Anti-TNF therapy: Safety aspects of taking the risk. Autoimmun. Rev. 2011, 10, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 44, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Perez, R.; Almoguera, B.; Cabaleiro, T.; Hakonarson, H.; Abad-Santos, F. Association between genetic polymorphisms and response to anti-TNFs in patients with inflammatory bowel disease. Int. J. Mol. Sci. 2016, 17, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, T.W.; Matheeuwsen, M.; Lonnkvist, M.H.; Parker, C.E.; Wildenberg, M.E.; Gecse, K.B.; D’Haens, G.R. Systematic review: Predictive biomarkers of therapeutic response in inflammatory bowel disease-personalised medicine in its infancy. Aliment. Pharmacol. Ther. 2018, 48, 1213–1231. [Google Scholar] [CrossRef]
- Aden, K.; Rehman, A.; Waschina, S.; Pan, W.-H.; Walker, A.; Lucio, M.; Nunez, A.M.; Bharti, R.; Zimmerman, J.; Bethge, J. Metabolic functions of gut microbes associate with efficacy of tumor necrosis factor antagonists in patients with inflammatory bowel diseases. Gastroenterology 2019, 157, 1279–1292.e1211. [Google Scholar] [CrossRef] [Green Version]
- Baert, F.J.; D’Haens, G.R.; Peeters, M.; Hiele, M.I.; Schaible, T.F.; Shealy, D.; Geboes, K.; Rutgeerts, P.J. Tumor necrosis factor α antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology 1999, 116, 22–28. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Scaldaferri, F.; Sgambato, A.; Rutella, S.; Cittadini, A.; Piqué, J.M.; Panes, J.; Katz, J.A.; Gasbarrini, A. TNF-α blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: A novel anti-inflammatory mechanism of infliximab in Crohn’s disease. J. Immunol. 2006, 176, 2617–2624. [Google Scholar] [CrossRef]
- Busquets, D.; Mas-de-Xaxars, T.; López-Siles, M.; Martínez-Medina, M.; Bahí, A.; Sàbat, M.; Louvriex, R.; Miquel-Cusachs, J.O.; Garcia-Gil, J.L.; Aldeguer, X. Anti-tumour necrosis factor treatment with adalimumab induces changes in the microbiota of Crohn’s disease. J. Crohns Colitis 2015, 9, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Rajca, S.; Grondin, V.; Louis, E.; Vernier-Massouille, G.; Grimaud, J.-C.; Bouhnik, Y.; Laharie, D.; Dupas, J.-L.; Pillant, H.; Picon, L. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 978–986. [Google Scholar] [PubMed]
- Kolho, K.-L.; Korpela, K.; Jaakkola, T.; Pichai, M.V.; Zoetendal, E.G.; Salonen, A.; De Vos, W.M. Fecal microbiota in pediatric inflammatory bowel disease and its relation to inflammation. Am. J. Gastroenterol. 2015, 110, 921. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis 2019, 13, 144–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.B.; Lee, K.-M.; Lee, J.M.; Chung, Y.Y.; Sung, H.J.; Paik, C.N.; Chung, W.C.; Jung, J.-H.; Choi, H.J. Correlation between histological activity and endoscopic, clinical, and serologic activities in patients with ulcerative colitis. Gastroenterol. Res. Pract. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Papamichael, K.; Gazouli, M.; Karakoidas, C.; Panayotou, I.; Roma-Giannikou, E.; Mantzaris, G.J. Association of TNF and FcγRΙΙΙA gene polymorphisms with differential response to infliximab in a Greek cohort of Crohn’s disease patients. Ann. Gastroenterol. 2011, 24, 35. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.-J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cao, K.-A.; Costello, M.-E.; Lakis, V.A.; Bartolo, F.; Chua, X.-Y.; Brazeilles, R.; Rondeau, P. mixMC: A multivariate statistical framework to gain insight into Microbial Communities. PLoS ONE 2016, 11, e0160169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodini, R.J.; Dowd, S.E.; Chamberlin, W.M.; Galandiuk, S.; Davis, B.; Glassing, A. Microbial population differentials between mucosal and submucosal intestinal tissues in advanced Crohn’s disease of the ileum. PLoS ONE 2015, 10, e0134382. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Rausch, P.; Wang, J.; Skieceviciene, J.; Kiudelis, G.; Bhagalia, K.; Amarapurkar, D.; Kupcinskas, L.; Schreiber, S.; Rosenstiel, P. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 2016, 65, 238–248. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.Z.; He, Y.; Yang, Y.; Liu, L.; Lin, Q.; Nie, Y.; Li, M.; Zhi, F.; Liu, S. Gut microbiota offers universal biomarkers across ethnicity in inflammatory bowel disease diagnosis and infliximab response prediction. MSystems 2018, 3, e00188-17. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.T.; Amos, G.C.; Murphy, A.R.; Murch, S.; Wellington, E.M.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef]
- Tang, M.S.; Poles, J.; Leung, J.M.; Wolff, M.J.; Davenport, M.; Lee, S.C.; Lim, Y.A.; Chua, K.H.; Loke, P.n.; Cho, I. Inferred metagenomic comparison of mucosal and fecal microbiota from individuals undergoing routine screening colonoscopy reveals similar differences observed during active inflammation. Gut Microbes 2015, 6, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Durbán, A.; Abellán, J.J.; Jiménez-Hernández, N.; Ponce, M.; Ponce, J.; Sala, T.; D’Auria, G.; Latorre, A.; Moya, A. Assessing gut microbial diversity from feces and rectal mucosa. Microb. Ecol. 2011, 61, 123–133. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Neurath, M.F.; Wirtz, S. The intestinal microbiota in inflammatory bowel disease. ILAR J. 2015, 56, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Andoh, A.; Sugimoto, M. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; Da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. J. Crohns Colitis 2015, 10, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Kiely, C.J.; Pavli, P.; O’brien, C.L. The microbiome of translocated bacterial populations in patients with and without inflammatory bowel disease. Intern. Med. J. 2018, 48, 1346–1354. [Google Scholar] [CrossRef]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef]
- Zitvogel, L.; Daillere, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef]
- Markou, P.; Apidianakis, Y. Pathogenesis of intestinal Pseudomonas aeruginosa infection in patients with cancer. Front. Cell. Infect. Microbiol. 2014, 3, 115. [Google Scholar] [CrossRef]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.; Nezi, L.; Reuben, A.; Andrews, M.; Karpinets, T.; Prieto, P.; Vicente, D.; Hoffman, K.; Wei, S. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.K.; Ding, T.; Koumpouras, C.; Telesco, S.E.; Monast, C.; Das, A.; Brodmerkel, C.; Schloss, P.D. Fecal microbiota signatures are associated with response to ustekinumab therapy among crohn’s disease patients. mBio 2018, 9, e02120-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Yang, L.; Cui, Z.; Zheng, J.; Huang, J.; Zhao, Q.; Su, Z.; Wang, M.; Zhang, W.; Liu, J.; et al. Anti-TNF-alpha therapy alters the gut microbiota in proteoglycan-induced ankylosing spondylitis in mice. MicrobiologyOpen 2019, 8, e927. [Google Scholar] [CrossRef] [PubMed]
- Picchianti-Diamanti, A.; Panebianco, C.; Salemi, S.; Sorgi, M.L.; Di Rosa, R.; Tropea, A.; Sgrulletti, M.; Salerno, G.; Terracciano, F.; D’Amelio, R.; et al. Analysis of gut microbiota in rheumatoid arthritis patients: Disease-related dysbiosis and modifications induced by etanercept. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.I.; Li, J.R.; Liu, C.C.; Liu, P.Y.; Chen, H.H. An association of gut microbiota with different phenotypes in chinese patients with rheumatoid arthritis. J. Clin. Med. 2019, 8, 1770. [Google Scholar] [CrossRef] [Green Version]
- Braun-Moscovici, Y.; Markovits, D.; Zinder, O.; Schapira, D.; Rozin, A.; Ehrenburg, M.; Dain, L.; Hoffer, E.; Nahir, A.M.; Balbir-Gurman, A. Anti-cyclic citrullinated protein antibodies as a predictor of response to anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis. J. Rheumatol. 2006, 33, 497–500. [Google Scholar]
- Lopetuso, L.; Gerardi, V.; Papa, V.; Scaldaferri, F.; Rapaccini, G.; Gasbarrini, A.; Papa, A. Can we predict the efficacy of anti-TNF-α agents? Int. J. Mol. Sci. 2017, 18, 1973. [Google Scholar] [CrossRef]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6, e000246. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Jones-Hall, Y.L.; Nakatsu, C.H. The intersection of TNF, IBD and the microbiome. Gut Microbes 2016, 7, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Zhu, C.; Quan, Y.; Yang, J.; Yuan, W.; Yang, Z.; Wu, S.; Luo, W.; Tan, B.; Wang, X. Insights into Roseburia intestinalis which alleviates experimental colitis pathology by inducing anti-inflammatory responses. J. Gastroenterol. Hepatol. 2018, 33, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420. [Google Scholar] [CrossRef] [PubMed]
- Toedter, G.; Li, K.; Marano, C.; Ma, K.; Sague, S.; Huang, C.C.; Song, X.Y.; Rutgeerts, P.; Baribaud, F. Gene expression profiling and response signatures associated with differential responses to infliximab treatment in ulcerative colitis. Am. J. Gastroenterol. 2011, 106, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Arijs, I.; De Hertogh, G.; Machiels, K.; Van Steen, K.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Quintens, R.; Van Assche, G.; Vermeire, S.; et al. Mucosal gene expression of cell adhesion molecules, chemokines, and chemokine receptors in patients with inflammatory bowel disease before and after infliximab treatment. Am. J. Gastroenterol. 2011, 106, 748–761. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Krause, P.; Zahner, S.P.; Kim, G.; Shaikh, R.B.; Steinberg, M.W.; Kronenberg, M. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology 2014, 146, 1752–1762.e1754. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef]
- McNamee, E.N.; Masterson, J.C.; Veny, M.; Collins, C.B.; Jedlicka, P.; Byrne, F.R.; Ng, G.Y.; Rivera-Nieves, J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 2015, 97, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Rocamora-Reverte, L.; Tuzlak, S.; von Raffay, L.; Tisch, M.; Fiegl, H.; Drach, M.; Reichardt, H.M.; Villunger, A.; Tischner, D.; Wiegers, G.J. Glucocorticoid receptor-deficient foxp3(+) regulatory T cells fail to control experimental inflammatory bowel disease. Front. Immunol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Atreya, R.; Neurath, M.F. Chemokines in inflammatory bowel diseases. Dig. Dis. 2010, 28, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Banks, C.; Bateman, A.; Payne, R.; Johnson, P.; Sheron, N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J. Pathol. 2003, 199, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, E.; Dobre, M.; Manuc, T.E.; Becheanu, G.; Tieranu, C.G.; Ionescu, E.M.; Manuc, M. Mucosal gene expression changes induced by anti-TNF treatment in inflammatory bowel disease patients. Drug Dev. Res. 2019, 80, 831–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.L.; Muehlbauer, A.L.; Alazizi, A.; Burns, M.B.; Findley, A.; Messina, F.; Gould, T.J.; Cascardo, C.; Pique-Regi, R.; Blekhman, R.; et al. Gut microbiota has a widespread and modifiable effect on host gene regulation. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro-Blanch, J.; Yanes, O. Epigenetic regulation at the interplay between gut microbiota and host metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Hasler, R.; Sheibani-Tezerji, R.; Sinha, A.; Barann, M.; Rehman, A.; Esser, D.; Aden, K.; Knecht, C.; Brandt, B.; Nikolaus, S.; et al. Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut 2017, 66, 2087–2097. [Google Scholar] [CrossRef]
- Magnusson, M.K.; Strid, H.; Sapnara, M.; Lasson, A.; Bajor, A.; Ung, K.A.; Ohman, L. Anti-TNF therapy response in patients with ulcerative colitis is associated with colonic antimicrobial peptide expression and microbiota composition. J. Crohns Colitis 2016, 10, 943–952. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Kerry, R.G.; Patra, J.K.; Gouda, S.; Park, Y.; Shin, H.-S.; Das, G. Benefaction of probiotics for human health: A review. J. Food Drug Anal. 2018, 26, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Taur, Y.; Coyte, K.; Schluter, J.; Robilotti, E.; Figueroa, C.; Gjonbalaj, M.; Littmann, E.R.; Ling, L.; Miller, L.; Gyaltshen, Y. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci. Transl. Med. 2018, 10, eaap9489. [Google Scholar] [CrossRef] [Green Version]
- Suez, J.; Zmora, N.; Zilberman-Schapira, G.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.-Z.; Federici, S. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 2018, 174, 1406–1423.e1416. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dovrolis, N.; Michalopoulos, G.; Theodoropoulos, G.E.; Arvanitidis, K.; Kolios, G.; Sechi, L.A.; Eliopoulos, A.G.; Gazouli, M. The Interplay between Mucosal Microbiota Composition and Host Gene-Expression is Linked with Infliximab Response in Inflammatory Bowel Diseases. Microorganisms 2020, 8, 438. https://doi.org/10.3390/microorganisms8030438
Dovrolis N, Michalopoulos G, Theodoropoulos GE, Arvanitidis K, Kolios G, Sechi LA, Eliopoulos AG, Gazouli M. The Interplay between Mucosal Microbiota Composition and Host Gene-Expression is Linked with Infliximab Response in Inflammatory Bowel Diseases. Microorganisms. 2020; 8(3):438. https://doi.org/10.3390/microorganisms8030438
Chicago/Turabian StyleDovrolis, Nikolas, George Michalopoulos, George E. Theodoropoulos, Kostantinos Arvanitidis, George Kolios, Leonardo A. Sechi, Aristidis G. Eliopoulos, and Maria Gazouli. 2020. "The Interplay between Mucosal Microbiota Composition and Host Gene-Expression is Linked with Infliximab Response in Inflammatory Bowel Diseases" Microorganisms 8, no. 3: 438. https://doi.org/10.3390/microorganisms8030438
APA StyleDovrolis, N., Michalopoulos, G., Theodoropoulos, G. E., Arvanitidis, K., Kolios, G., Sechi, L. A., Eliopoulos, A. G., & Gazouli, M. (2020). The Interplay between Mucosal Microbiota Composition and Host Gene-Expression is Linked with Infliximab Response in Inflammatory Bowel Diseases. Microorganisms, 8(3), 438. https://doi.org/10.3390/microorganisms8030438