The Association between Gut Microbiota and Uremia of Chronic Kidney Disease

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Clinical Information on Study Participants
2.3. Stool DNA Extraction and MiSeq Sequencing
2.4. Sequence Data Analysis
2.5. Serum Metabolite Analysis
2.6. Statistical Analysis
3. Results
3.1. Comparisons of Baseline Characteristics and Serum Uremic Metabolites according to CKD Group
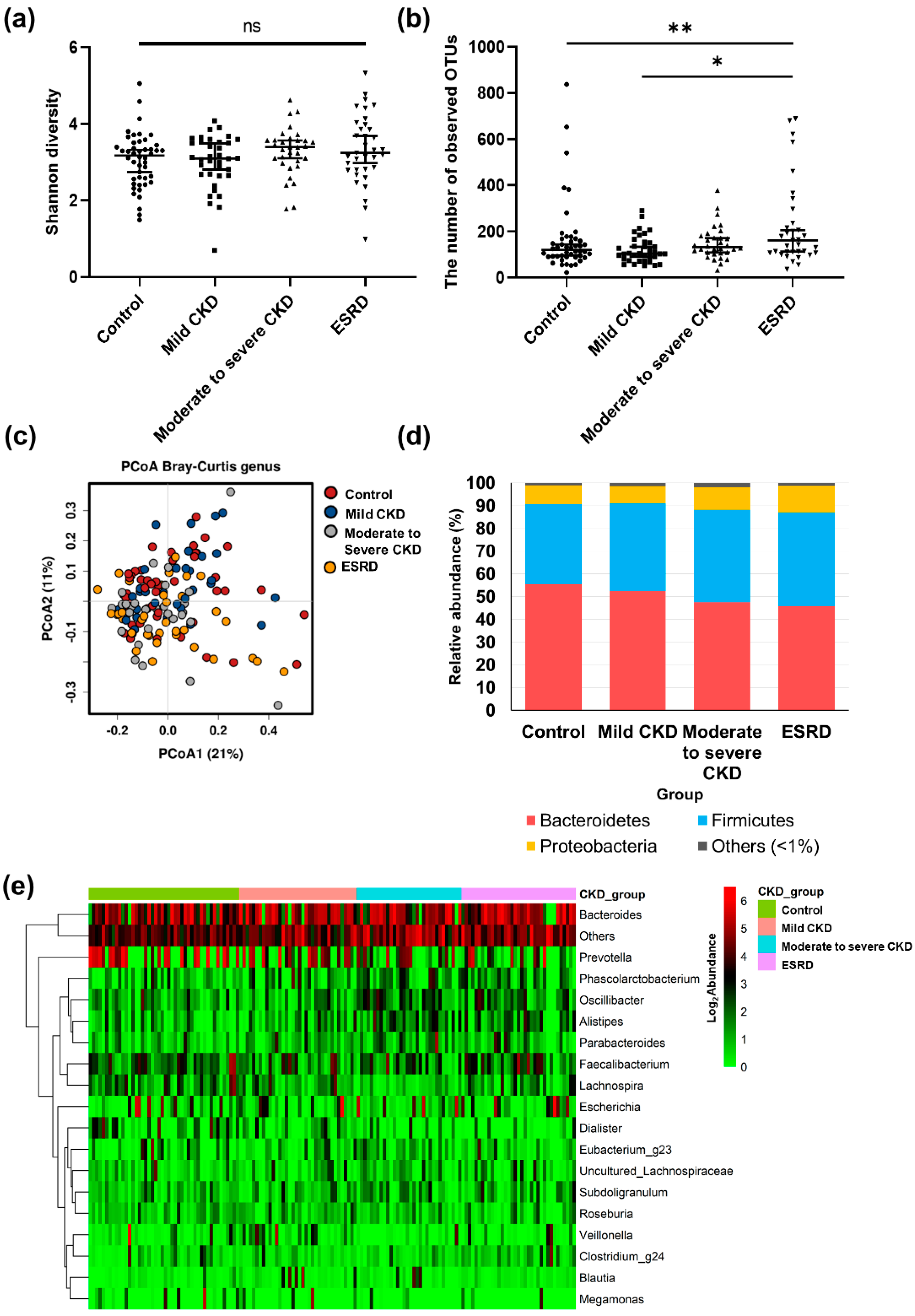
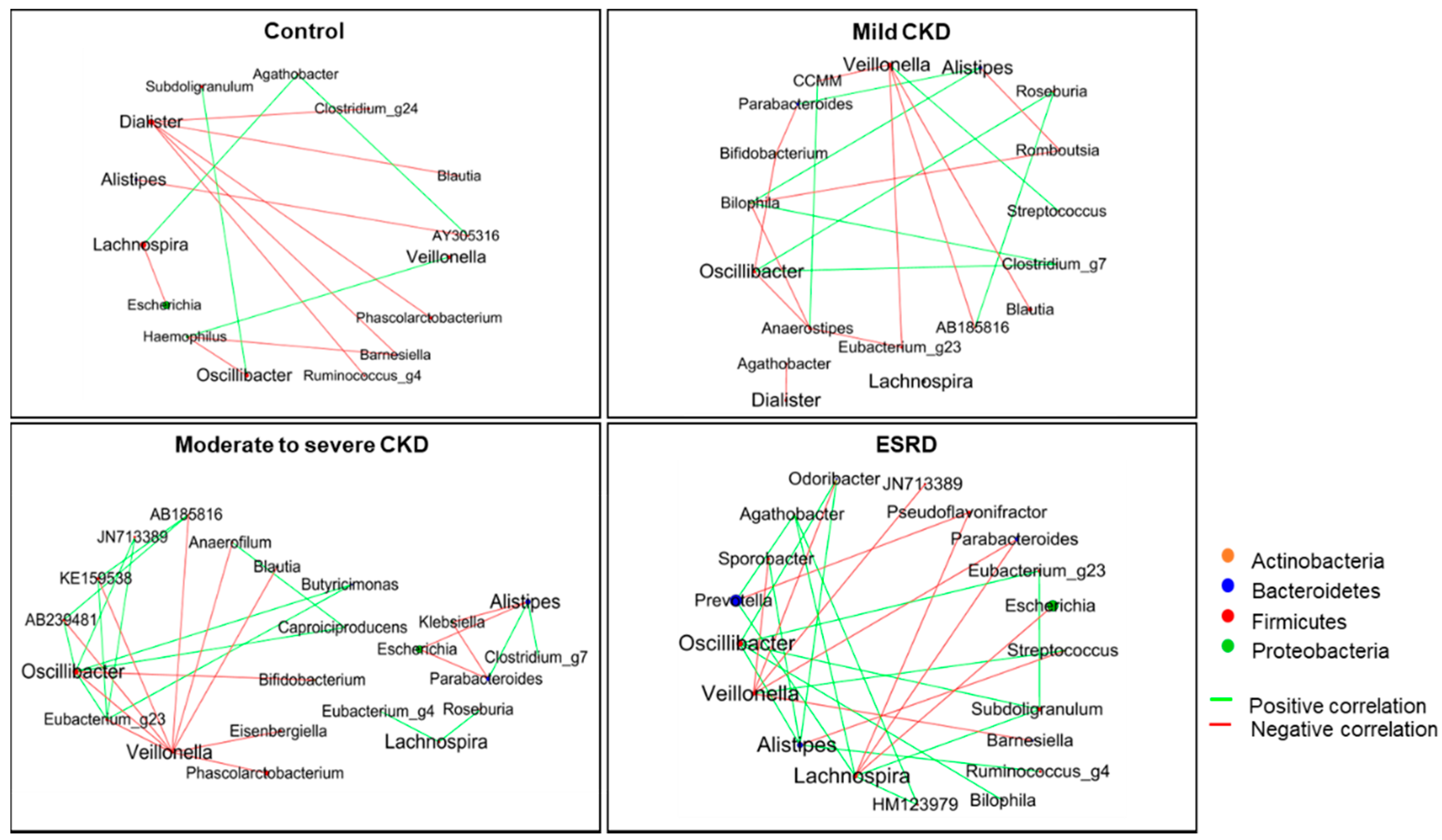
3.2. Differences in Microbiota Composition according to CKD Group
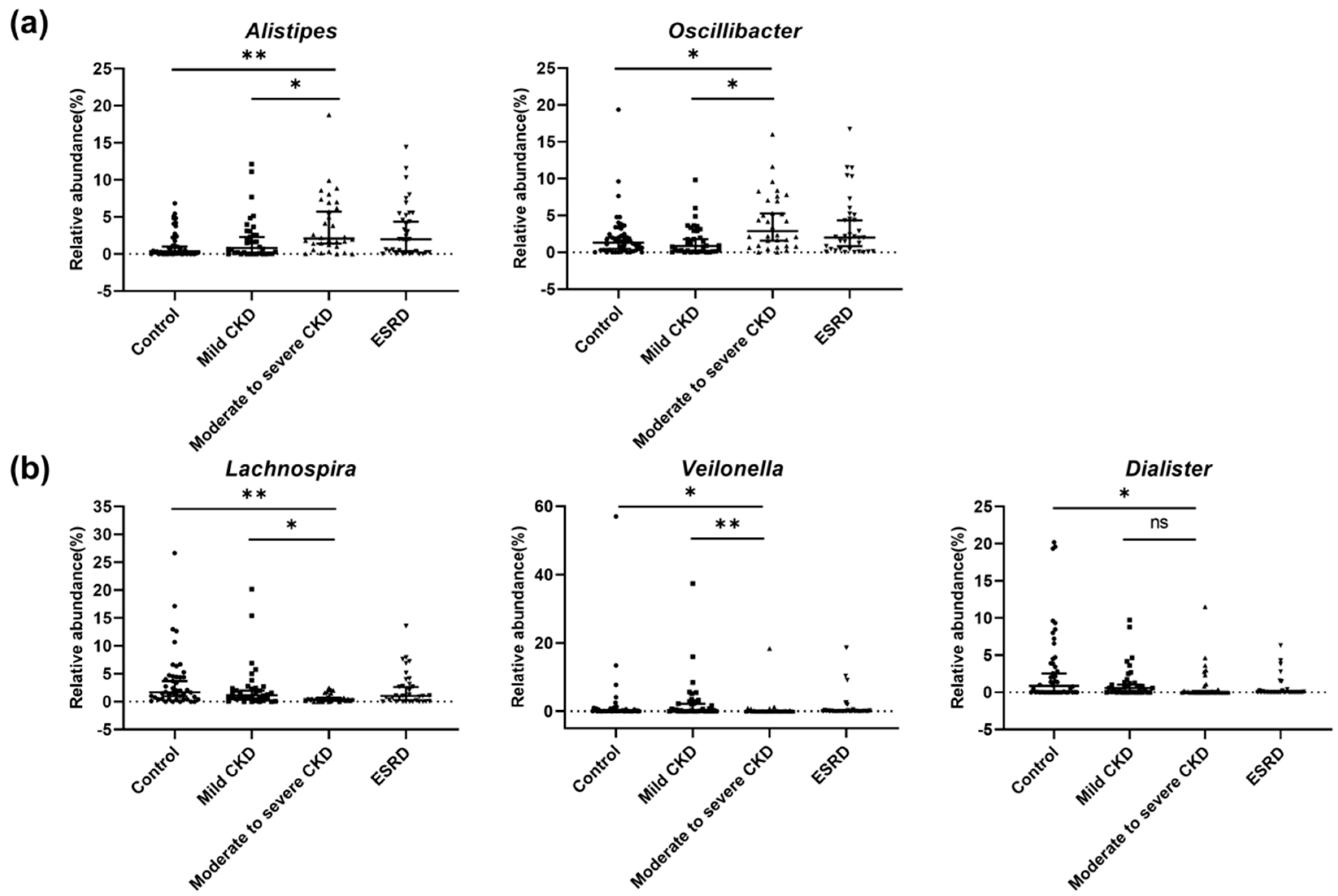
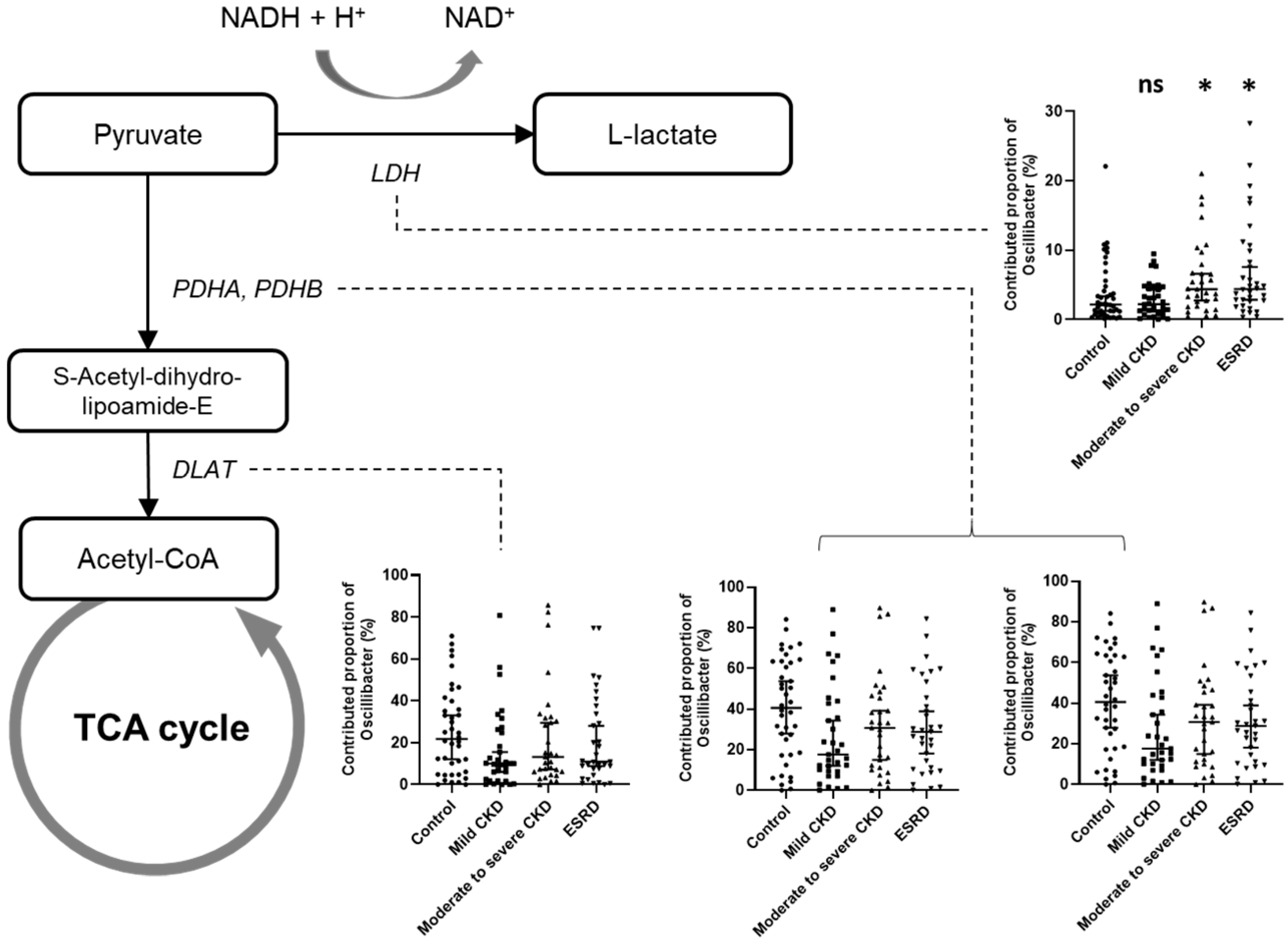
3.3. Microbiota-Related Uremic Toxins
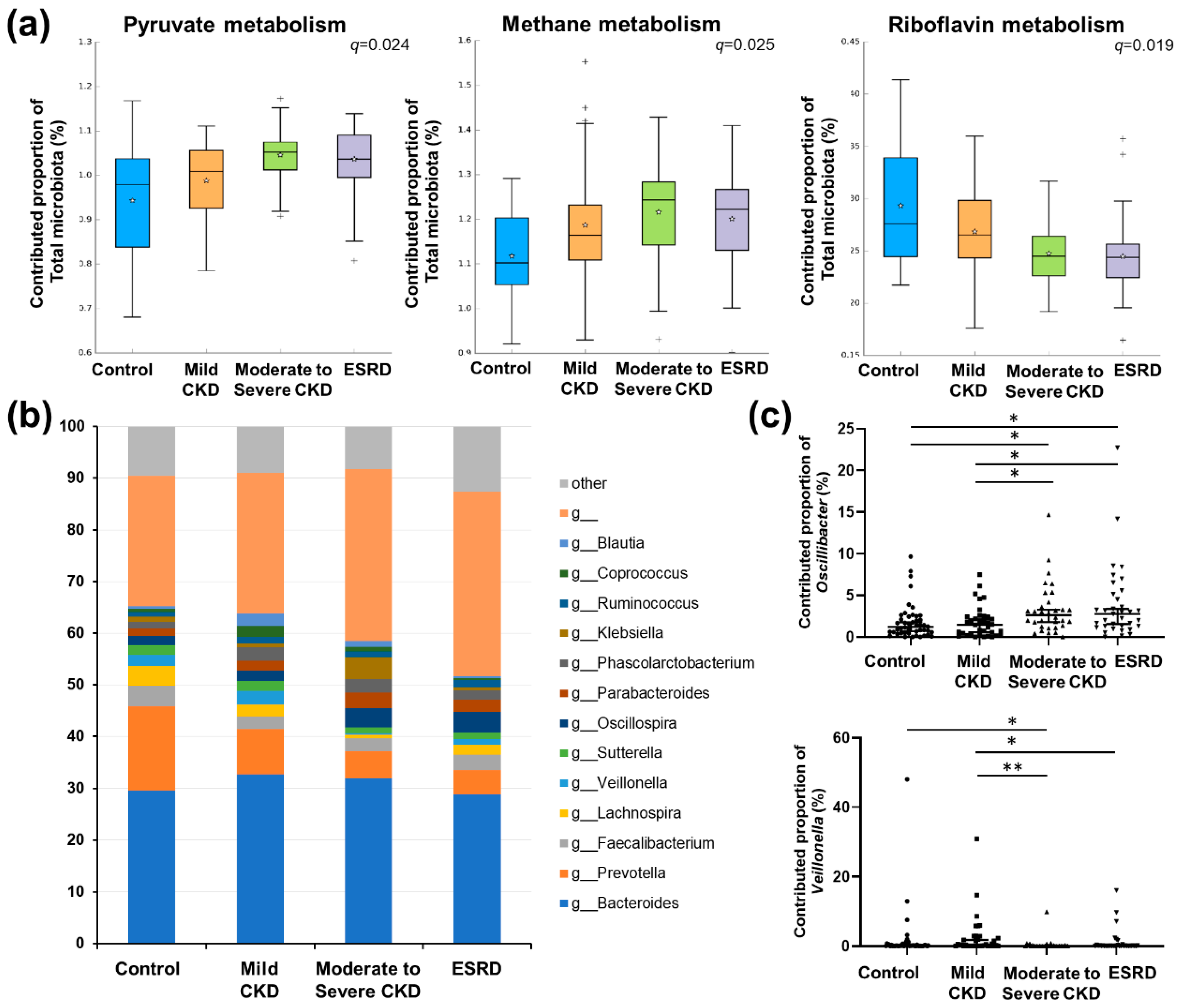
3.4. Predicted Functional Analysis of Gut Microbiota among the CKD Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Glendinning, L.; Free, A. Supra-organismal interactions in the human intestine. Front. Cell. Infect. Microbiol. 2014, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoodpoor, F.; Rahbar Saadat, Y.; Barzegari, A.; Ardalan, M.; Zununi Vahed, S. The impact of gut microbiota on kidney function and pathogenesis. Biomed. Pharmacother. 2017, 93, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- de Almeida Duarte, J.B.; de Aguilar-Nascimento, J.E.; Nascimento, M.; Nochi, R.J., Jr. Bacterial translocation in experimental uremia. Urol. Res. 2004, 32, 266–270. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.D.; Sirich, T.L.; Meyer, T.W. Uremic Toxin Clearance and Cardiovascular Toxicities. Toxins (Basel) 2018, 10, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.Y.; Hsu, H.J.; Tsai, C.J.; Tzen, C.Y.; Wang, Y.C.; Lin, C.Y.; Wu, M.S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Meert, N.; Schepers, E.; Glorieux, G.; Argiles, A.; Brunet, P.; Cohen, G.; Drueke, T.; Mischak, H.; Spasovski, G.; et al. Review on uraemic solutes II—Variability in reported concentrations: Causes and consequences. Nephrol. Dial. Transplant. 2007, 22, 3115–3121. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [Green Version]
- Melamed, M.L.; Plantinga, L.; Shafi, T.; Parekh, R.; Meyer, T.W.; Hostetter, T.H.; Coresh, J.; Powe, N.R. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol. 2013, 14, 134. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T. Role of indoxyl sulfate in the progression of chronic kidney disease and cardiovascular disease: Experimental and clinical effects of oral sorbent AST-120. Ther. Apher. Dial. 2011, 15, 120–124. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins (Basel) 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.T. Gut commensals make choline too. Nat. Microbiol. 2019, 4, 4–5. [Google Scholar] [CrossRef]
- Barrows, I.R.; Ramezani, A.; Raj, D.S. Gut Feeling in AKI: The Long Arm of Short-Chain Fatty Acids. J. Am. Soc. Nephrol. 2015, 26, 1755–1757. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Gong, J.; Cottrill, M.; Yu, H.; de Lange, C.; Burton, J.; Topp, E. Evaluation of QIAamp DNA Stool Mini Kit for ecological studies of gut microbiota. J. Microbiol. Methods 2003, 54, 13–20. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work, G. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef] [Green Version]
- Kanemitsu, Y.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Saigusa, D.; Mukawa, C.; Tachikawa, T.; Abe, T.; Tomioka, Y. Simultaneous quantitative analysis of uremic toxins by LC-MS/MS with a reversed-phase/cation-exchange/anion-exchange tri-modal mixed-mode column. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1068–1069, 1–8. [Google Scholar] [CrossRef]
- Shu, C.; Chen, X.; Xia, T.; Zhang, F.; Gao, S.; Chen, W. LC-MS/MS method for simultaneous determination of serum p-cresyl sulfate and indoxyl sulfate in patients undergoing peritoneal dialysis. Biomed. Chromatogr. 2016, 30, 1782–1788. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Mondot, S.; Kang, S.; Furet, J.P.; Aguirre de Carcer, D.; McSweeney, C.; Morrison, M.; Marteau, P.; Dore, J.; Leclerc, M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm. Bowel Dis. 2011, 17, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkila, J.; Marchesi, J.R.; Smidt, H.; de Vos, W.M.; Ross, R.P.; O’Toole, P.W. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, Y.Y.; Ha, C.W.; Campbell, C.R.; Mitchell, A.J.; Dinudom, A.; Oscarsson, J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 2012, 7, e34233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Hu, J.; Iragavarapu, S.; Nadkarni, G.N.; Huang, R.; Erazo, M.; Bao, X.; Verghese, D.; Coca, S.; Ahmed, M.K.; Peter, I. Location-Specific Oral Microbiome Possesses Features Associated With CKD. Kidney Int. Rep. 2018, 3, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Petriz, B.A.; Castro, A.P.; Almeida, J.A.; Gomes, C.P.; Fernandes, G.R.; Kruger, R.H.; Pereira, R.W.; Franco, O.L. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genom. 2014, 15, 511. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Ilhan, Z.E.; Marcus, A.K.; Kang, D.W.; Rittmann, B.E.; Krajmalnik-Brown, R. pH-Mediated Microbial and Metabolic Interactions in Fecal Enrichment Cultures. mSphere 2017, 2, e00047-17. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Control | Mild CKD | Moderate to Severe CKD | ESRD | |
|---|---|---|---|---|---|
| Total N = 149 | N = 46 | N = 36 | N = 32 | N = 35 | p |
| Clinical parameters | |||||
| Age (y) | 47.0 ± 10.8 | 49.8 ± 15.1 | 52.4 ± 11.9 | 48.9 ± 12.2 | 0.251 |
| Male sex (%) | 16 (34.8) | 21 (58.3) | 17 (53.1) | 15 (42.9) | 0.15 |
| Body mass index (%) | 23.3 ± 3.0 | 24.6 ± 3.5 | 23.6 ± 3.2 | 21.8 ± 4.3 | 0.003 |
| Diabetes mellitus (%) | 0 (0) | 3 (8.3) | 6 (18.8) | 6 (17.1) | 0.02 |
| Hypertension (%) | 3 (6.5) | 21 (58.3) | 22 (68.8) | 26 (74.3) | <0.001 |
| Blood urea nitrogen (mg/dL) | 12.1 ± 2.9 | 14.4 ± 3.9 | 43.7 ± 25.7 | 45.8 ± 15.8 | <0.001 |
| Serum creatinine (mg/dL) | 0.7 ± 0.2 | 0.8 ± 0.2 | 3.8 ± 2.5 | 7.8 ± 2.6 | <0.001 |
| CKD-EPI eGFR (mL/min/1.73 m2) | 101.6 ± 19.0 | 98.3 ± 26.1 | 25.2 ± 17.2 | 7.2 ± 2.5 | <0.001 |
| Urine RBC (number /HPF) | <0.001 | ||||
| 0 | 24 (52.2) | 4 (11.1) | 7 (21.9) | NA | |
| 1–4 | 17 (37.0) | 7 (19.4) | 9 (28.1) | NA | |
| 5≤ | 5 (10.9) | 25 (69.4) | 16 (50.0) | NA | |
| Urine protein/creatinine ratio | 0.05 ± 0.03 | 3.6 ± 3.4 | 3.3 ± 3.4 | NA | <0.001 |
| Plasma hemoglobin (g/dL) | 13.8 ± 1.3 | 12.9 ± 1.6 | 11.1 ± 2.0 | 10.3 ± 1.6 | <0.001 |
| Anemia (%) | 3 (6.5) | 13 (36.1) | 24 (75.0) | 30 (85.7) | <0.001 |
| Serum albumin (mg/dL) | 4.4 ± 0.3 | 3.5 ± 0.7 | 3.8 ± 0.5 | 3.8 ± 0.4 | <0.001 |
| Serum C-reactive protein (mg/dL) | 0.1 ± 0.4 | 0.2 ± 0.3 | 0.7 ± 1.3 | 0.3 ± 0.8 | 0.321 |
| Etiology of CKD (biopsy proven/clinical diagnosis) | <0.001 | ||||
| Diabetes mellitus | NA | 0 | 4 (0/4) | 6 (0/6) | |
| Hypertension | NA | 0 | 1 (1/0) | 1 (0/1) | |
| Glomerulonephritis | NA | 35 (35/0) | 21 (18/3) | 14 (4/10) | |
| Polycystic kidney | NA | 0 | 4 (0/4) | 3 (0/3) | |
| Others | NA | 1 | 2 | 11 | |
| Serum uremic metabolites | |||||
| P-cresyl sulfate (ug/mL) | 9.5 ± 10.8 | 7.00 ± 8.7 | 63.2 ± 56.0 | 111.6 ± 87.0 | <0.001 |
| P-cresyl glucuronide * (ng/mL) | 18.2 ± 18.0 | 19.8 ± 19.3 | 114.5 ± 110.1 | 746.7 ± 880.5 | <0.001 |
| Indoxyl sulfate (ug/mL) | 0.7 ± 0.4 | 0.7 ± 0.6 | 7.3 ± 7.6 | 26.0 ± 17.8 | <0.001 |
| TMAO (ug/mL) | 0.6 ± 1.1 | 0.8 ± 1.2 | 4.9 ± 5.9 | 13.9 ± 17.4 | <0.001 |
| Predictors | Regression Coefficient | Standard Error | Adjusted R2 | p | FDR |
|---|---|---|---|---|---|
| p-cresyl sulfate (log) | |||||
| Alistipes | 0.207 | 0.050 | 0.100 | <0.001 | <0.001 |
| Oscillibacter | 0.238 | 0.045 | 0.155 | <0.001 | <0.001 |
| Lachnospira | –0.105 | 0.043 | 0.033 | 0.016 | 0.039 |
| Veillonella | –0.079 | 0.027 | 0.050 | 0.004 | 0.014 |
| Subdoligranulum | 0.234 | 0.085 | 0.042 | 0.007 | 0.023 |
| Megamonas | –0.092 | 0.036 | 0.036 | 0.012 | 0.034 |
| p-cresyl glucuronate (log) * | |||||
| Prevotella | –0.023 | 0.009 | 0.034 | 0.014 | 0.062 |
| Alistipes | 0.189 | 0.057 | 0.062 | 0.001 | 0.010 |
| Oscillibacter | 0.213 | 0.053 | 0.094 | < 0.001 | 0.001 |
| Lachnospira | –0.104 | 0.049 | 0.024 | 0.034 | 0.103 |
| Subdoligranulum | 0.224 | 0.097 | 0.028 | 0.023 | 0.081 |
| Indoxyl sulfate (log) | |||||
| Alistipes | 0.126 | 0.043 | 0.048 | 0.004 | 0.035 |
| Oscillibacter | 0.112 | 0.041 | 0.043 | 0.007 | 0.037 |
| Lachnospira | –0.091 | 0.036 | 0.034 | 0.014 | 0.058 |
| Subdoligranulum | 0.177 | 0.073 | 0.032 | 0.016 | 0.058 |
| TMAO (log) | |||||
| Prevotella | –0.016 | 0.007 | 0.027 | 0.026 | 0.080 |
| Alistipes | 0.104 | 0.046 | 0.027 | 0.026 | 0.080 |
| Oscillibacter | 0.145 | 0.042 | 0.067 | 0.001 | 0.006 |
| Lachnospira | –0.087 | 0.039 | 0.026 | 0.027 | 0.080 |
| Dialister | –0.107 | 0.043 | 0.034 | 0.014 | 0.080 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.E.; Kim, H.-E.; Park, J.I.; Cho, H.; Kwak, M.-J.; Kim, B.-Y.; Yang, S.H.; Lee, J.P.; Kim, D.K.; Joo, K.W.; et al. The Association between Gut Microbiota and Uremia of Chronic Kidney Disease. Microorganisms 2020, 8, 907. https://doi.org/10.3390/microorganisms8060907
Kim JE, Kim H-E, Park JI, Cho H, Kwak M-J, Kim B-Y, Yang SH, Lee JP, Kim DK, Joo KW, et al. The Association between Gut Microbiota and Uremia of Chronic Kidney Disease. Microorganisms. 2020; 8(6):907. https://doi.org/10.3390/microorganisms8060907
Chicago/Turabian StyleKim, Ji Eun, Hyo-Eun Kim, Ji In Park, Hyunjeong Cho, Min-Jung Kwak, Byung-Yong Kim, Seung Hee Yang, Jung Pyo Lee, Dong Ki Kim, Kwon Wook Joo, and et al. 2020. "The Association between Gut Microbiota and Uremia of Chronic Kidney Disease" Microorganisms 8, no. 6: 907. https://doi.org/10.3390/microorganisms8060907
APA StyleKim, J. E., Kim, H. -E., Park, J. I., Cho, H., Kwak, M. -J., Kim, B. -Y., Yang, S. H., Lee, J. P., Kim, D. K., Joo, K. W., Kim, Y. S., Kim, B. -S., & Lee, H. (2020). The Association between Gut Microbiota and Uremia of Chronic Kidney Disease. Microorganisms, 8(6), 907. https://doi.org/10.3390/microorganisms8060907