
Myxobacterial Genomics and Post-Genomics: A Review of Genome Biology, Genome Sequences and Related ‘Omics Studies



Abstract
:
1. Myxobacterial Genomics
1.1. The Genome Sequence of M. xanthus DK1622
1.2. Other Early Genome Sequences
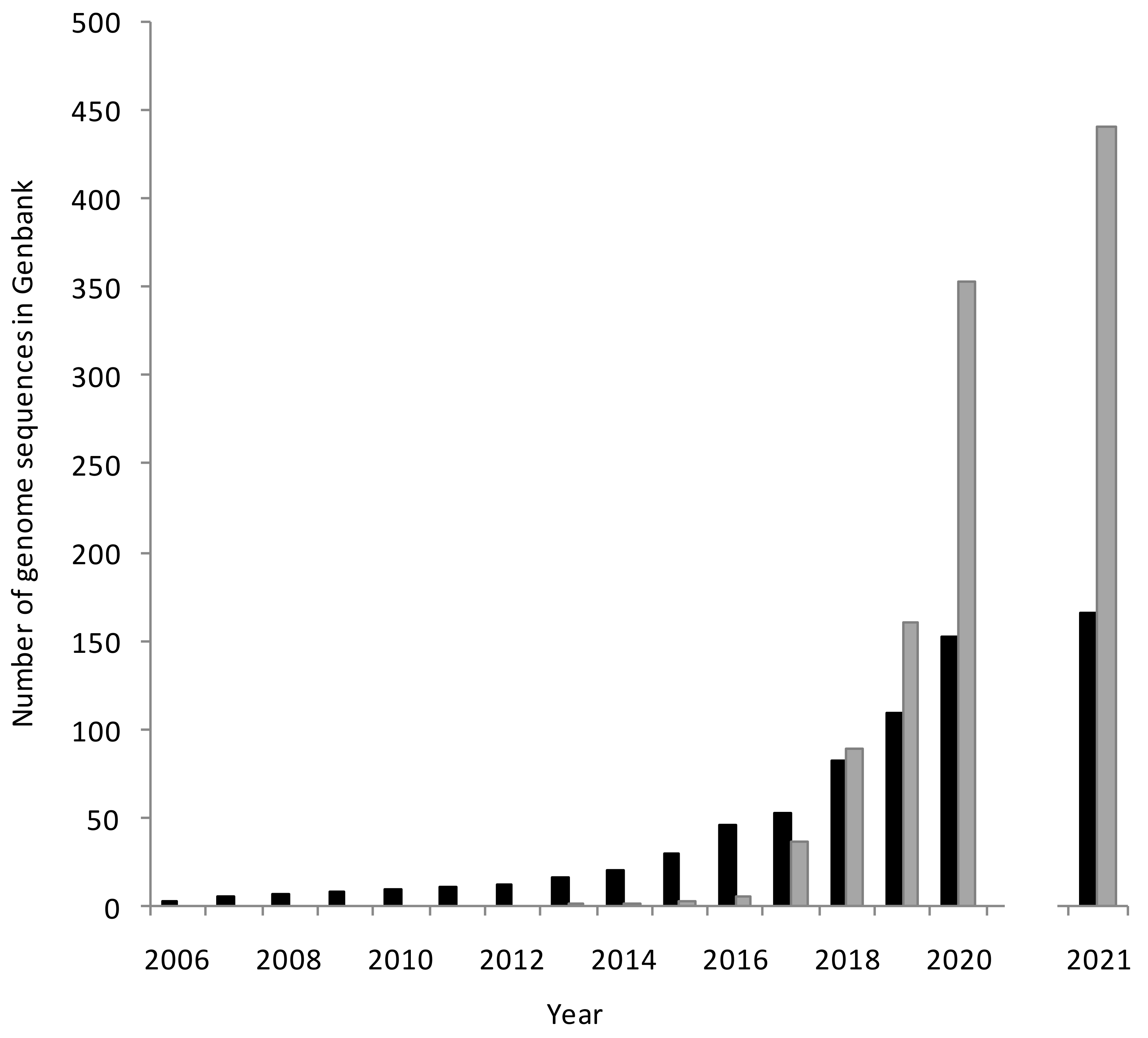
1.3. Expanding Coverage and Increasing Depth
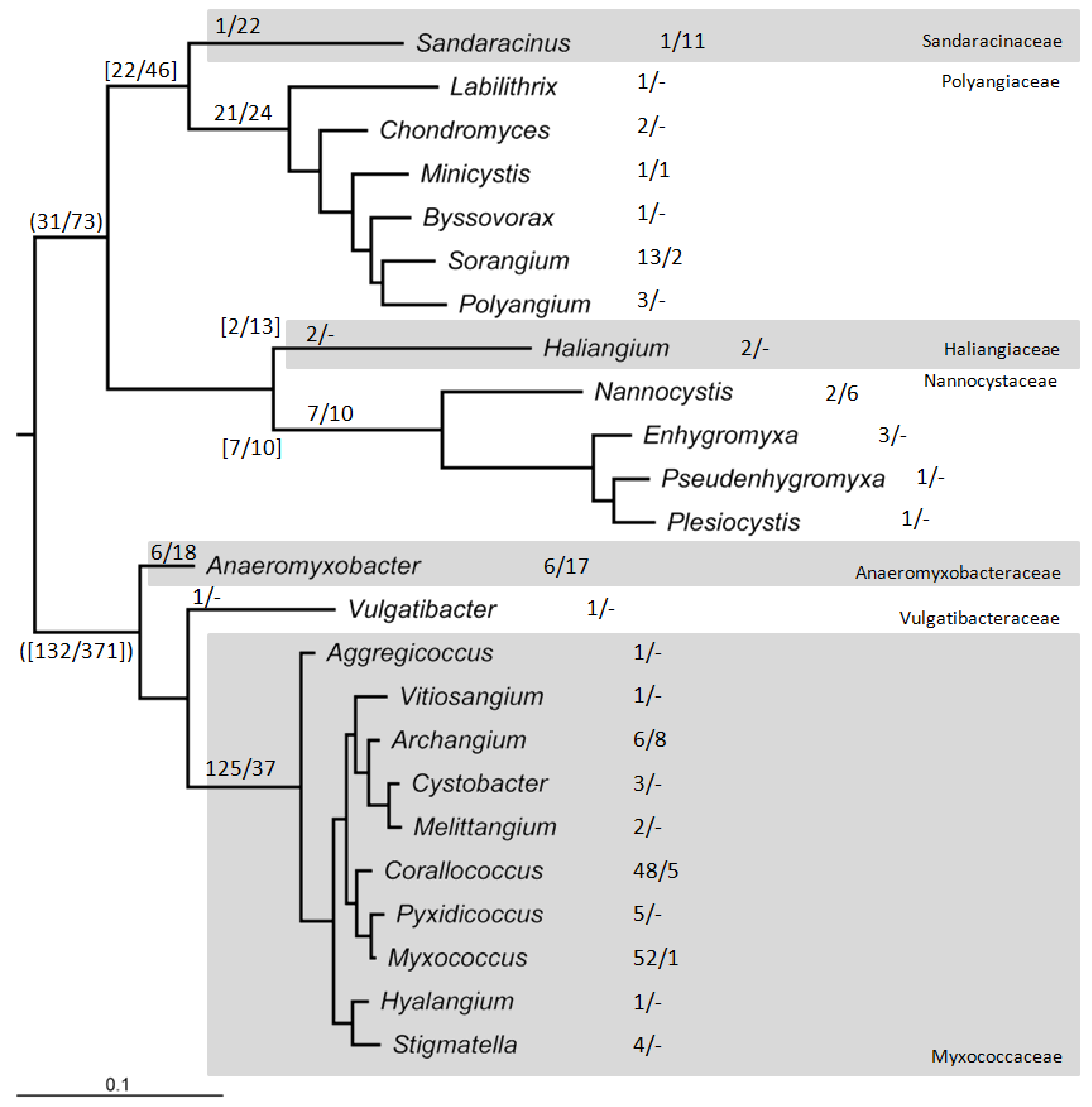
1.4. Genome Sequences and Myxobacterial Classification
2. Myxobacterial Genome Biology
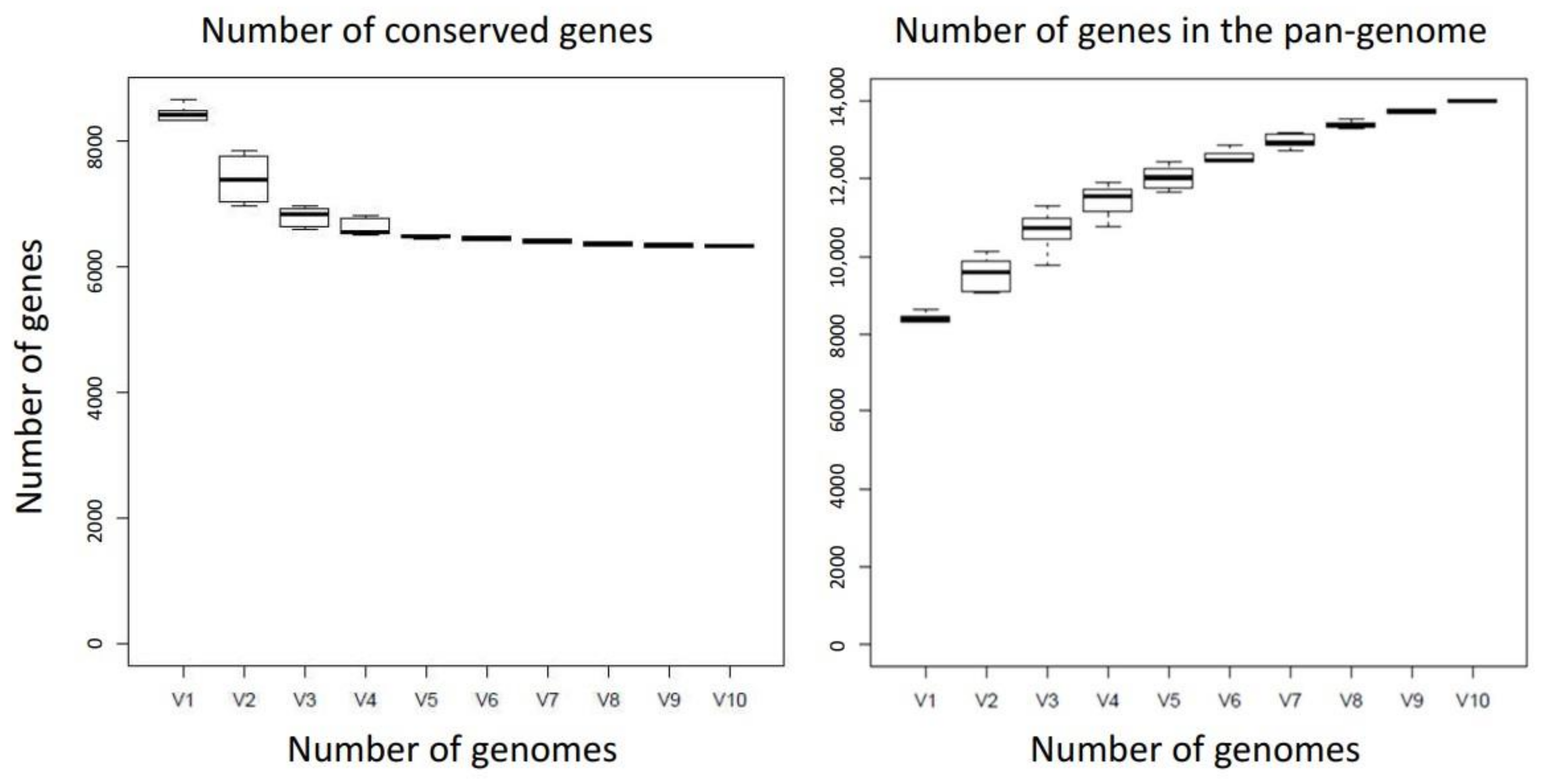
2.1. Pan-Genomics
2.2. Comparative Studies—Gene Repertoires
2.3. Genome Organisation
2.4. Genome Evolution
2.5. Comparative Studies—Evolution of Specific Myxobacterial Systems
3. Myxobacterial Post-Genomics
3.1. Molecular Genetics
3.2. Transcriptomics
3.3. Proteomics
3.4. Metabolomics and Interactomics
4. Perspectives
- Single-cell transcriptomics will be combined with advanced imaging techniques and single-cell tracking to investigate the epigenetic effects of life history on individuals in a population.
- MAGs will direct efforts to define and cultivate novel taxa which are currently unculturable.
- Genome editing and/or recombineering will be used to produce high-throughput combinatorial gene deletions for investigations into gene function.
- Single amplified genomes will provide insights into evolutionary processes within natural populations.
- Proteomics methods will be used holistically to assess post-translational modifications, particularly those associated with epigenetic regulation of metabolism and signalling.
- Artificial intelligence will be used to integrate multi-omic data and physiological data into systems models and to generate hypotheses for testing.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A
References
- Yang, Z.; Higgs, P.I. Myxobacteria: Genomics, Cellular and Molecular Biology; Caister Academic Press: Norfolk, UK, 2014. [Google Scholar]
- Whitworth, D.E. Myxobacteria: Multicellularity and Differentiation; ASM Press: Washington, DC, USA, 2008. [Google Scholar]
- Findlay, B.L. The Chemical Ecology of Predatory Soil Bacteria. ACS Chem. Biol. 2016, 11, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Furness, E.; Whitworth, D.E.; Zwarycz, A. Predatory Interactions between Myxobacteria and Their Prey. In The Ecology of Predation at the Microscale; Jurkevitch, E., Mitchell, R.J., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–36. [Google Scholar]
- Petters, S.; Groß, V.; Söllinger, A.; Pichler, M.; Reinhard, A.; Bengtsson, M.M.; Urich, T. The soil microbial food web revisited: Predatory myxobacteria as keystone taxa? ISME J. 2021, 15, 2665–2675. [Google Scholar] [CrossRef]
- Muñoz-Dorado, J.; Torres, F.J.M.; García-Bravo, E.; Moraleda-Munoz, A.; Pérez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc. Natl. Acad. Sci. USA 1979, 76, 5952–5956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroos, L. Highly Signal-Responsive Gene Regulatory Network Governing Myxococcus Development. Trends Genet. 2017, 33, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.A.; Garza, A.G. Genetic Tools for Studying Myxococcus xanthus Biology. In Myxobacteria: Multicellularity and Differentiation; Whitworth, D.E., Ed.; ASM Press: Washington, DC, USA, 2014; pp. 491–501. [Google Scholar]
- Kuspa, A.; Vollrath, D.; Cheng, Y.; Kaiser, D. Physical mapping of the Myxococcus xanthus genome by random cloning in yeast artificial chromosomes. Proc. Natl. Acad. Sci. USA 1989, 86, 8917–8921. [Google Scholar] [CrossRef] [Green Version]
- Zusman, D.R.; Krotoski, D.M.; Cumsky, M. Chromosome replication in Myxococcus xanthus. J. Bacteriol. 1978, 133, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Ordal, E.J. Deoxyribonucleic Acid Homology in Bacterial Taxonomy: Effect of Incubation Temperature on Reaction Specificity. J. Bacteriol. 1968, 95, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, D.; Manoil, C.; Dworkin, M. Myxobacteria: Cell Interactions, Genetics, and Development. Annu. Rev. Microbiol. 1979, 33, 595–639. [Google Scholar] [CrossRef]
- Inouye, S.; Ike, Y.; Inouye, M. Tandem repeat of the genes for protein S, a development-specific protein of Myxococcus xanthus. J. Biol. Chem. 1983, 258, 38–40. [Google Scholar] [CrossRef]
- Romeo, J.M.; Esmon, B.; Zusman, D.R. Nucleotide sequence of the myxobacterial hemagglutinin gene contains four homologous domains. Proc. Natl. Acad. Sci. USA 1986, 83, 6332–6336. [Google Scholar] [CrossRef] [Green Version]
- Oden, S.; Brocchieri, L. Quantitative frame analysis and the annotation of GC-rich (and other) prokaryotic genomes. An application to Anaeromyxobacter dehalogenans. Bioinformatics 2015, 31, 3254–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.H.; Wagner, R.D.; Arakaki, A.K.; Skolnick, J.; Kirby, J.R.; Shimkets, L.J.; Sanford, R.A.; Löffler, F.E. The Mosaic Genome of Anaeromyxobacter dehalogenans Strain 2CP-C Suggests an Aerobic Common Ancestor to the Delta-Proteobacteria. PLoS ONE 2008, 3, e2103. [Google Scholar] [CrossRef] [Green Version]
- Goldman, B.S.; Nierman, W.C.; Kaiser, D.; Slater, S.C.; Durkin, A.S.; Eisen, J.A.; Ronning, C.M.; Barbazuk, W.; Blanchard, M.; Field, C.; et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. USA 2006, 103, 15200–15205. [Google Scholar] [CrossRef] [Green Version]
- Huntley, S.; Wuichet, K.; Søgaard-Andersen, L. Genome evolution and content in the myxobacteria. In Myxobacteria: Genomics, Cellular and Molecular Biology; Yang, Z., Higgs, P.I., Eds.; Caister Academic Press: Norfolk, UK, 2014; pp. 31–50. [Google Scholar]
- Gophna, U.; Charlebois, R.L.; Doolittle, W.F. Ancient lateral gene transfer in the evolution of Bdellovibrio bacteriovorus. Trends Microbiol. 2006, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Schneiker, S.; Perlova, O.; Kaiser, O.; Gerth, K.; Alici, A.; O Altmeyer, M.; Bartels, D.; Bekel, T.; Beyer, S.; Bode, E.; et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat. Biotechnol. 2007, 25, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, A.P.; Kaiser, D. Nutrition of Myxococcus xanthus, a fruiting myxobacterium. J. Bacteriol. 1978, 133, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, N.; Daum, C.; Lang, E.; Abt, B.; Kopitz, M.; Saunders, E.; Lapidus, A.; Lucas, S.; Del Rio, T.G.; Nolan, M.; et al. Complete genome sequence of Haliangium ochraceum type strain (SMP-2T). Stand. Genom. Sci. 2010, 2, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Huntley, S.; Hamann, N.; Wegener-Feldbrugge, S.; Treuner-Lange, A.; Kube, M.; Reinhardt, R.; Klages, S.; Müller, R.; Ronning, C.M.; Nierman, W.C.; et al. Comparative Genomic Analysis of Fruiting Body Formation in Myxococcales. Mol. Biol. Evol. 2010, 28, 1083–1097. [Google Scholar] [CrossRef] [Green Version]
- Huntley, S.; Zhang, Y.; Treuner-Lange, A.; Kneip, S.; Sensen, C.W.; Søgaard-Andersen, L. Complete Genome Sequence of the Fruiting Myxobacterium Corallococcus coralloides DSM 2259. J. Bacteriol. 2012, 194, 3012–3013. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-F.; Li, X.; Liu, H.; Liu, X.; Han, K.; Wu, Z.-H.; Hu, W.; Li, F.-F.; Li, Y.-Z. Genome Sequence of the Halotolerant Marine Bacterium Myxococcus fulvus HW-1. J. Bacteriol. 2011, 193, 5015–5016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, K.I.; Stechling, M.; Wink, J.; Wilharm, E.; Stadler, M. Comparison of myxobacterial diversity and evaluation of isolation success in two niches: Kiritimati Island and German compost. MicrobiologyOpen 2016, 5, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Copeland, A.; Lucas, S.; Lapidus, A.; Barry, K.; del Rio, T.G.; Dalin, E.; Tice, H.; Pitluck, S.; Sims, D.; et al. Complete Genome Sequence of Anaeromyxobacter sp. Fw109-5, an Anaerobic, Metal-Reducing Bacterium Isolated from a Contaminated Subsurface Environment. Genome Announc. 2015, 3, e01449-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segerman, B. The Most Frequently Used Sequencing Technologies and Assembly Methods in Different Time Segments of the Bacterial Surveillance and RefSeq Genome Databases. Front. Cell. Infect. Microbiol. 2020, 10, 527102. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, P.; Morphew, R.; Whitworth, D.E. Genome Sequencing and Pan-Genome Analysis of 23 Corallococcus spp. Strains Reveal Unexpected Diversity, with Particular Plasticity of Predatory Gene Sets. Front. Microbiol. 2018, 9, 3187. [Google Scholar] [CrossRef] [Green Version]
- Pogorevc, D.; Panter, F.; Schillinger, C.; Jansen, R.; Wenzel, S.C.; Müller, R. Production optimization and biosynthesis revision of corallopyronin A, a potent anti-filarial antibiotic. Metab. Eng. 2019, 55, 201–211. [Google Scholar] [CrossRef]
- Wielgoss, S.; Wolfensberger, R.; Sun, L.; Fiegna, F.; Velicer, G.J. Social genes are selection hotspots in kin groups of a soil microbe. Science 2019, 363, 1342–1345. [Google Scholar] [CrossRef]
- Livingstone, P.; Morphew, R.; Whitworth, D.E. Myxobacteria Are Able to Prey Broadly upon Clinically-Relevant Pathogens, Exhibiting a Prey Range Which Cannot Be Explained by Phylogeny. Front. Microbiol. 2017, 8, 1593. [Google Scholar] [CrossRef] [Green Version]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [Green Version]
- Mohr, K.I.; Wolf, C.; Nübel, U.; Szafrańska, A.K.; Steglich, M.; Hennessen, F.; Gemperlein, K.; Kampfer, P.; Martin, K.; Müller, R.; et al. A polyphasic approach leads to seven new species of the cellulose-decomposing genus Sorangium, Sorangium ambruticinum sp. nov., Sorangium arenae sp. nov., Sorangium bulgaricum sp. nov., Sorangium dawidii sp. nov., Sorangium kenyense sp. nov., Sorangium orientale sp. nov. and Sorangium reichenbachii sp. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 3576–3586. [Google Scholar] [CrossRef]
- Garcia, R.; Gerth, K.; Stadler, M.; Dogma, I.J.; Müller, R. Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol. Phylogenet. Evol. 2010, 57, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Sutton, D.; Livingstone, P.; Furness, E.; Swain, M.T.; Whitworth, D.E. Genome-Wide Identification of Myxobacterial Predation Genes and Demonstration of Formaldehyde Secretion as a Potentially Predation-Resistant Trait of Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2650. [Google Scholar] [CrossRef] [PubMed]
- Ahearne, A.; Albataineh, H.; Dowd, S.; Stevens, D. Assessment of Evolutionary Relationships for Prioritization of Myxobacteria for Natural Product Discovery. Microorganisms 2021, 9, 1376. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, P.G.; Ingleby, O.; Girdwood, S.; Cookson, A.R.; Morphew, R.; Whitworth, D.E. Predatory Organisms with Untapped Biosynthetic Potential: Descriptions of Novel Corallococcus Species C. aberystwythensis sp. nov., C. carmarthensis sp. nov., C. exercitus sp. nov., C. interemptor sp. nov., C. llansteffanensis sp. nov., C. praedator sp. nov., C. sicarius sp. nov. and C. terminator sp. nov. Appl. Environ. Microbiol. 2020, 86, e01931-19. [Google Scholar] [CrossRef]
- Chambers, J.; Sparks, N.; Sydney, N.; Livingstone, P.G.; Cookson, A.R.; E Whitworth, D. Comparative Genomics and Pan-Genomics of the Myxococcaceae, including a Description of Five Novel Species: Myxococcus eversor sp. nov., Myxococcus llanfairpwllgwyngyllgogerychwyrndrobwllllantysiliogogogochensis sp. nov., Myxococcus vastator sp. nov., Pyxidicoccus caerfyrddinensis sp. nov. and Pyxidicoccus trucidator sp. nov. Genome Biol. Evol. 2020, 12, 2289–2302. [Google Scholar] [CrossRef]
- Murray, A.E.; Freudenstein, J.; Gribaldo, S.; Hatzenpichler, R.; Hugenholtz, P.; Kampfer, P.; Konstantinidis, K.T.; Lane, C.E.; Papke, R.; Parks, D.H.; et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 2020, 5, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Chuvochina, M.; Pelikan, C.; Parks, D.H.; Yilmaz, P.; Wagner, M.; Loy, A.; Naganuma, T.; Nakai, R.; Whitman, W.B.; et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int. J. Syst. Evol. Microbiol. 2020, 70, 5972–6016. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Wielgoss, S.; Didelot, X.; Chaudhuri, R.; Liu, X.; Weedall, G.; Velicer, G.J.; Vos, M. A barrier to homologous recombination between sympatric strains of the cooperative soil bacterium Myxococcus xanthus. ISME J. 2016, 10, 2468–2477. [Google Scholar] [CrossRef] [Green Version]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.; Angiuoli, S.V.; Oggioni, M.; Hotopp, J.C.D.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef] [Green Version]
- Argemi, X.; Matelska, D.; Ginalski, K.; Riegel, P.; Hansmann, Y.; Bloom, J.; Pestel-Caron, M.; Dahyot, S.; Lebeurre, J.; Prévost, G. Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genom. 2018, 19, 621. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Zwarycz, A.S.; Livingstone, P.; Whitworth, D.E. Within-species variation in OMV cargo proteins: The Myxococcus xanthus OMV pan-proteome. Mol. Omics 2020, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Gemperlein, K.; Zaburannyi, N.; Garcia, R.; La Clair, J.J.; Müller, R. Metabolic and Biosynthetic Diversity in Marine Myxobacteria. Mar. Drugs 2018, 16, 314. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Moghaddam, J.A.; Crüsemann, M.; Alanjary, M.; Harms, H.; Dávila-Céspedes, A.; Blom, J.; Poehlein, A.; Ziemert, N.; König, G.M.; Schäberle, T.F. Analysis of the Genome and Metabolome of Marine Myxobacteria Reveals High Potential for Biosynthesis of Novel Specialized Metabolites. Sci. Rep. 2018, 8, 16600. [Google Scholar] [CrossRef] [PubMed]
- Khatri, Y.; Hannemann, F.; Perlova, O.; Müller, R.; Bernhardt, R. Investigation of cytochromes P450 in myxobacteria: Excavation of cytochromes P450 from the genome of Sorangium cellulosum So ce56. FEBS Lett. 2011, 585, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Burgard, C.; Zaburannyi, N.; Nadmid, S.; Maier, J.; Jenke-Kodama, H.; Luxenburger, E.; Bernauer, H.S.; Wenzel, S.C. Genomics-Guided Exploitation of Lipopeptide Diversity in Myxobacteria. ACS Chem. Biol. 2017, 12, 779–786. [Google Scholar] [CrossRef]
- Gregory, K.; Salvador, L.A.; Akbar, S.; Adaikpoh, B.; Stevens, D.C. Survey of Biosynthetic Gene Clusters from Sequenced Myxobacteria Reveals Unexplored Biosynthetic Potential. Microorganisms 2019, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Hug, J.J.; Panter, F.; Krug, D.; Müller, R. Genome mining reveals uncommon alkylpyrones as type III PKS products from myxobacteria. J. Ind. Microbiol. Biotechnol. 2019, 46, 319–334. [Google Scholar] [CrossRef]
- Perez, J.; Castaneda-Garcia, A.; Jenke-Kodama, H.; Muller, R.; Munoz-Dorado, J. Eukaryotic-like protein kinases in the prokaryotes and the myxobacterial kinome. Proc. Natl. Acad. Sci. USA 2008, 105, 15950–15955. [Google Scholar] [CrossRef] [Green Version]
- Treuner-Lange, A. The Phosphatomes of the Multicellular Myxobacteria Myxococcus xanthus and Sorangium cellulosum in Comparison with Other Prokaryotic Genomes. PLoS ONE 2010, 5, e11164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, D.E.; Cock, P.J.A. Two-component systems of the myxobacteria: Structure, diversity and evolutionary relationships. Microbiology 2008, 154, 360–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, D.E.; Cock, P.J.A. Myxobacterial Two-Component Systems. In Myxobacteria: Multicellularity and Differentiation; Whitworth, D.E., Ed.; ASM Press: Washington, DC, USA, 2008; pp. 169–189. [Google Scholar]
- Sharma, G.; Khatri, I.; Subramanian, S. Comparative Genomics of Myxobacterial Chemosensory Systems. J. Bacteriol. 2018, 200, e00620-17. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, D. Signaling in myxobacteria. Annu. Rev. Microbiol. 2004, 58, 75–98. [Google Scholar] [CrossRef]
- Whitworth, D.E.; Zwarycz, A. A Genomic Survey of Signalling in the Myxococcaceae. Microorganisms 2020, 8, 1739. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Parkhill, J. Comparative Genomic Structure of Prokaryotes. Annu. Rev. Genet. 2004, 38, 771–791. [Google Scholar] [CrossRef] [Green Version]
- Khedkar, S.; Seshasayee, A.S.N. Comparative Genomics of Interreplichore Translocations in Bacteria: A Measure of Chromosome Topology? G3 2016, 6, 1597–1606. [Google Scholar] [CrossRef] [Green Version]
- Mackiewicz, P. Where does bacterial replication start? Rules for predicting the oriC region. Nucleic Acids Res. 2004, 32, 3781–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Zhang, C.-T. Origins of Replication in Sorangium cellulosum and Microcystis aeruginosa. DNA Res. 2008, 15, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, D.E.; Swain, M.T. A survey of non-coding RNAs in the social and predatory myxobacterium Myxococcus xanthus DK1622. Mol. Omics 2020, 16, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.R.; Whitworth, D.E. Correlations between the role, sequence conservation, genomic location and severity of phenotype in myxobacterial developmental genes. FEMS Microbiol. Lett. 2010, 312, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, P.; Hanquier, J.; Jaoua, S.; Reichenbach, H.; Guespin-Michel, J.F. Utilization of IncP-1 Plasmids as Vectors for Transposon Mutagenesis in Myxobacteria. Microbiology 1988, 134, 2889–2895. [Google Scholar] [CrossRef] [Green Version]
- Magrini, V.; Creighton, C.; Youderian, P. Site-Specific Recombination of Temperate Myxococcus xanthus Phage Mx8: Genetic Elements Required for Integration. J. Bacteriol. 1999, 181, 4050–4061. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.-Y.; Zhong, L.; Shen, M.-J.; Xia, Z.; Cheng, Q.-X.; Sun, X.; Zhao, G.-P.; Li, Y.; Qin, Z.-J. Discovery of the Autonomously Replicating Plasmid pMF1 from Myxococcus fulvus and Development of a Gene Cloning System in Myxococcus xanthus. Appl. Environ. Microbiol. 2008, 74, 1980–1987. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-J.; Liu, Y.; Zhang, Z.; Chen, X.-J.; Gong, Y.; Li, Y. A Post-segregational Killing Mechanism for Maintaining Plasmid PMF1 in Its Myxococcus fulvus Host. Front. Cell. Infect. Microbiol. 2018, 8, 274. [Google Scholar] [CrossRef]
- Chen, X.-J.; Han, K.; Feng, J.; Zhuo, L.; Li, Y.-J.; Li, Y.-Z. The complete genome sequence and analysis of a plasmid-bearing myxobacterial strain Myxococcus fulvus 124B02 (M 206081). Stand. Genom. Sci. 2016, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.E. Genome-wide analysis of myxobacterial two-component systems: Genome relatedness and evolutionary changes. BMC Genom. 2015, 16, 780. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, S.J.; Thrash, C.; Temperton, B. Implications of streamlining theory for microbial ecology. ISME J. 2014, 8, 1553–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltrus, D.A. Exploring the costs of horizontal gene transfer. Trends Ecol. Evol. 2013, 28, 489–495. [Google Scholar] [CrossRef]
- Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb. Genom. 2018, 4, e000168. [Google Scholar] [CrossRef]
- Kumbhar, C.; Mudliar, P.; Bhatia, L.; Kshirsagar, A.; Watve, M. Widespread predatory abilities in the genus Streptomyces. Arch. Microbiol. 2014, 196, 235–248. [Google Scholar] [CrossRef]
- Nair, R.R.; Vasse, M.; Wielgoss, S.; Sun, L.; Yu, Y.-T.N.; Velicer, G.J. Bacterial predator-prey coevolution accelerates genome evolution and selects on virulence-associated prey defences. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Parish, J.H. Transfer of Drug Resistance to Myxococcus from Bacteria Carrying Drug-resistance Factors. J. Gen. Microbiol. 1975, 87, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hu, W.; Lux, R.; He, X.; Li, Y.; Shi, W. Natural Transformation of Myxococcus xanthus. J. Bacteriol. 2011, 193, 2122–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasse, M.; Wielgoss, S. Bacteriophages of Myxococcus xanthus, a Social Bacterium. Viruses 2018, 10, 374. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, C.N.; Cao, P.; Conklin, A.; Finkelstein, H.; Hayes, C.S.; Wall, D. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife 2017, 6, 29397. [Google Scholar] [CrossRef]
- Goldman, B.; Bhat, S.; Shimkets, L.J. Genome Evolution and the Emergence of Fruiting Body Development in Myxococcus xanthus. PLoS ONE 2007, 2, e1329. [Google Scholar] [CrossRef] [Green Version]
- Desmond, E.; Gribaldo, S. Phylogenomics of Sterol Synthesis: Insights into the Origin, Evolution, and Diversity of a Key Eukaryotic Feature. Genome Biol. Evol. 2009, 1, 364–381. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-C.K.; Griesenauer, B.; Yu, Y.-T.N.; Velicer, G.J. A recent evolutionary origin of a bacterial small RNA that controls multicellular fruiting body development. Mol. Phylogenet. Evol. 2014, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Wielgoss, S.; Lippert, G.; Velicer, G.J.; Kroos, L. devIIs an Evolutionarily Young Negative Regulator of Myxococcus xanthus Development. J. Bacteriol. 2015, 197, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Luciano, J.; Agrebi, R.; Le Gall, A.V.; Wartel, M.; Fiegna, F.; Ducret, A.; Brochier-Armanet, C.; Mignot, T. Emergence and Modular Evolution of a Novel Motility Machinery in Bacteria. PLoS Genet. 2011, 7, e1002268. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Burrows, L.L.; Singer, M. Diversity and Evolution of Myxobacterial Type IV Pilus Systems. Front. Microbiol. 2018, 9, 1630. [Google Scholar] [CrossRef]
- López-García, P.; Moreira, D. The Syntrophy hypothesis for the origin of eukaryotes revisited. Nat. Microbiol. 2020, 5, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S. Evidence for myxobacterial origin of eukaryotic defensins. Immunogenetics 2007, 59, 949–954. [Google Scholar] [CrossRef]
- Elías-Arnanz, M.; Padmanabhan, S.; Murillo, F.J. The regulatory action of the myxobacterial CarD/CarG complex: A bacterial enhanceosome? FEMS Microbiol. Rev. 2010, 34, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.S.; Jelsbak, L.; Jelsbak, L.; Welch, R.D.; Cummings, C.; Goldman, B.; Stark, E.; Slater, S.; Kaiser, D. σ 54 Enhancer Binding Proteins and Myxococcus xanthus Fruiting Body Development. J. Bacteriol. 2004, 186, 4361–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, D.E.; Millard, A.; Hodgson, D.A.; Hawkins, P.F. Protein-protein interactions between two-component system transmitter and receiver domains of Myxococcus xanthus. Proteomics 2008, 8, 1839–1842. [Google Scholar] [CrossRef]
- Vassallo, C.N.; Wall, D. Self-identity barcodes encoded by six expansive polymorphic toxin families discriminate kin in myxobacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 24808–24818. [Google Scholar] [CrossRef]
- Whitworth, D.E. Genomes and knowledge—A questionable relationship? Trends Microbiol. 2008, 16, 512–519. [Google Scholar] [CrossRef]
- Velicer, G.J.; Raddatz, G.; Keller, H.; Deiss, S.; Lanz, C.; Dinkelacker, I.; Schuster, S.C. Comprehensive mutation identification in an evolved bacterial cooperator and its cheating ancestor. Proc. Natl. Acad. Sci. USA 2006, 103, 8107–8112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Welch, R.D.; Jakobsen, J.S.; Slater, S.C.; Garza, A.G. Global Mutational Analysis of NtrC-Like Activators in Myxococcus xanthus: Identifying Activator Mutants Defective for Motility and Fruiting Body Development. J. Bacteriol. 2003, 185, 6083–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, D.E.; Bryan, S.J.; Berry, A.; McGowan, S.; Hodgson, D.A. Genetic Dissection of the Light-Inducible carQRS Promoter Region of Myxococcus xanthus. J. Bacteriol. 2004, 186, 7836–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, T.; Inouye, S.; Inouye, M. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene 1996, 183, 153–157. [Google Scholar] [CrossRef]
- Iniesta, A.A.; García-Heras, F.; Abellón-Ruiz, J.; García, A.G.; Elías-Arnanz, M. Two Systems for Conditional Gene Expression in Myxococcus xanthus Inducible by Isopropyl-β- d -Thiogalactopyranoside or Vanillate. J. Bacteriol. 2012, 194, 5875–5885. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.G.; Treuner-Lange, A.; Moraleda-Munoz, A.; García-Bravo, E.; García-Hernández, R.; Martínez-Cayuela, M.; Pérez, J.; Søgaard-Andersen, L.; Muñoz-Dorado, J. Comprehensive Set of Integrative Plasmid Vectors for Copper-Inducible Gene Expression in Myxococcus xanthus. Appl. Environ. Microbiol. 2012, 78, 2515–2521. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-J.; Singh, R.P.; Lan, X.; Zhang, C.-S.; Li, Y.-Z.; Li, Y.-Q.; Sheng, D.-H. Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System. Biomolecules 2018, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-J.; Wang, Y.; Li, Z.-F.; Gong, Y.; Zhang, P.; Hu, W.-C.; Sheng, D.-H.; Li, Y.-Z. Increasing on-target cleavage efficiency for CRISPR/Cas9-induced large fragment deletion in Myxococcus xanthus. Microb. Cell Factories 2017, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Peng, R.; Wang, Y.; Feng, W.-W.; Yue, X.-J.; Chen, J.-H.; Hu, X.-Z.; Li, Z.-F.; Sheng, D.-H.; Zhang, Y.-M.; Li, Y.-Z. CRISPR/dCas9-mediated transcriptional improvement of the biosynthetic gene cluster for the epothilone production in Myxococcus xanthus. Microb. Cell Factories 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Whitworth, D.E.; Holmes, A.B.; Irvine, A.G.; Hodgson, D.A.; Scanlan, D.J. Phosphate Acquisition Components of the Myxococcus xanthus Pho Regulon Are Regulated by both Phosphate Availability and Development. J. Bacteriol. 2008, 190, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- Kroos, L.; Kaiser, D. Construction of Tn5 lac, a transposon that fuses lacZ expression to exogenous promoters, and its introduction into Myxococcus xanthus. Proc. Natl. Acad. Sci. USA 1984, 81, 5816–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Mishima, Y.; Nakano, H.; Takegawa, K. An Adenylyl Cyclase, CyaA, of Myxococcus xanthus Functions in Signal Transduction during Osmotic Stress. J. Bacteriol. 2002, 184, 3578–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diodati, M.E.; Ossa, F.; Caberoy, N.B.; Jose, I.R.; Hiraiwa, W.; Igo, M.M.; Singer, M.; Garza, A.G. Nla18, a Key Regulatory Protein Required for Normal Growth and Development of Myxococcus xanthus. J. Bacteriol. 2006, 188, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, M.; Wegener-Feldbrügge, S.; Søgaard-Andersen, L. The Orphan Response Regulator DigR Is Required for Synthesis of Extracellular Matrix Fibrils in Myxococcus xanthus. J. Bacteriol. 2006, 188, 4384–4394. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.D.; Shebelut, C.W.; Jose, I.R.; Hodgson, D.A.; Whitworth, D.E.; Singer, M. The response regulator PhoP4 is required for late developmental events in Myxococcus xanthus. Microbiol. 2006, 152, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, S.V.; Wegener-Feldbrügge, S.; Søgaard-Andersen, L.; Velicer, G.J. Novel Transcriptome Patterns Accompany Evolutionary Restoration of Defective Social Development in the Bacterium Myxococcus xanthus. Mol. Biol. Evol. 2008, 25, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Bode, H.B.; Ring, M.W.; Schwär, G.; Altmeyer, M.O.; Kegler, C.; Jose, I.R.; Singer, M.; Müller, R. Identification of Additional Players in the Alternative Biosynthesis Pathway to Isovaleryl-CoA in the Myxobacterium Myxococcus xanthus. ChemBioChem 2009, 10, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wegener-Feldbrügge, S.; Huntley, S.; Hamann, N.; Hedderich, R.; Søgaard-Andersen, L. Bioinformatics and Experimental Analysis of Proteins of Two-Component Systems in Myxococcus xanthus. J. Bacteriol. 2008, 190, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.-D.; Treuner-Lange, A.; Heider, J.; Huntley, S.M.; I Higgs, P. Global transcriptome analysis of spore formation in Myxococcus xanthus reveals a locus necessary for cell differentiation. BMC Genom. 2010, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, G.; Dziewanowska, K.; Stone, H.; Settles, M.; Hartzell, P. Global analysis of phase variation in Myxococcus xanthus. Mol. Microbiol. 2011, 81, 784–804. [Google Scholar] [CrossRef] [Green Version]
- Croucher, N.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-P.; Yue, X.-J.; Han, K.; Li, Z.-F.; Zheng, L.-S.; Yi, X.-N.; Wang, H.-L.; Zhang, Y.-M.; Li, Y.-Z. Allopatric integrations selectively change host transcriptomes, leading to varied expression efficiencies of exotic genes in Myxococcus xanthus. Microb. Cell Factories 2015, 14, 105. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, P.; Millard, A.D.; Swain, M.T.; Whitworth, D.E. Transcriptional changes when Myxococcus xanthus preys on Escherichia coli suggest myxobacterial predators are constitutively toxic but regulate their feeding. Microb. Genom. 2018, 4, e000152. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Dorado, J.; Moraleda-Munoz, A.; Torres, F.J.M.; Contreras-Moreno, F.J.; Martin-Cuadrado, A.B.; Schrader, J.M.; I Higgs, P.; Pérez, J. Transcriptome dynamics of the Myxococcus xanthus multicellular developmental program. eLife 2019, 8, 50374. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Yao, A.I.; Smaldone, G.T.; Liang, J.; Long, M.; Facciotti, M.T.; Singer, M. Global gene expression analysis of the Myxococcus xanthus developmental time course. Genomics 2021, 113, 120–134. [Google Scholar] [CrossRef]
- Whitfield, D.L.; Sharma, G.; Smaldone, G.T.; Singer, M. Peripheral rods: A specialized developmental cell type in Myxococcus xanthus. Genomics 2020, 112, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Sheng, D.-H.; Wang, Y.; Wu, S.-G.; Duan, R.-Q.; Li, Y.-Z. The Regulation of LexA on UV-Induced SOS Response in Myxococcus xanthus Based on Transcriptome Analysis. J. Microbiol. Biotechnol. 2021, 31, 912–920. [Google Scholar] [CrossRef]
- Curtis, P.D.; Atwood, J.; Orlando, R.; Shimkets, L.J. Proteins Associated with the Myxococcus xanthus Extracellular Matrix. J. Bacteriol. 2007, 189, 7634–7642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahnt, J.; Aguiluz, K.; Koch, J.; Treuner-Lange, A.; Konovalova, A.; Huntley, S.; Hoppert, M.; Søgaard-Andersen, L.; Hedderich, R. Profiling the Outer Membrane Proteome during Growth and Development of the Social Bacterium Myxococcus xanthus by Selective Biotinylation and Analyses of Outer Membrane Vesicles. J. Proteome Res. 2010, 9, 5197–5208. [Google Scholar] [CrossRef]
- Bhat, S.; Zhu, X.; Patel, R.P.; Orlando, R.; Shimkets, L.J. Identification and Localization of Myxococcus xanthus Porins and Lipoproteins. PLoS ONE 2011, 6, e27475. [Google Scholar] [CrossRef]
- Evans, A.G.L.; Davey, H.; Cookson, A.; Currinn, H.; Cooke-Fox, G.; Stanczyk, P.J.; Whitworth, D.E. Predatory activity of Myxococcus xanthus outer-membrane vesicles and properties of their hydrolase cargo. Microbiology 2012, 158, 2742–2752. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.L.; Tengra, F.K.; Dutton, D.; Yan, J.; Andacht, T.M.; Coyne, L.; Windell, V.; Garza, A.G. Identification of Major Sporulation Proteins of Myxococcus xanthus Using a Proteomic Approach. J. Bacteriol. 2007, 189, 3187–3197. [Google Scholar] [CrossRef] [Green Version]
- Chao, T.; Kalinowski, J.; Nyalwidhe, J.; Hansmeier, N. Comprehensive proteome profiling of the Fe(III)-reducing myxobacterium Anaeromyxobacter dehalogenans 2CP-C during growth with fumarate and ferric citrate. Proteomics 2010, 10, 1673–1684. [Google Scholar] [CrossRef]
- Izzat, S.; Rachid, S.; Ajdidi, A.; El-Nakady, Y.A.; Liu, X.-X.; Ye, B.-C.; Müller, R. The ROK like protein of Myxococcus xanthus DK1622 acts as a pleiotropic transcriptional regulator for secondary metabolism. J. Biotechnol. 2020, 311, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schley, C.; Altmeyer, M.O.; Swart, R.; Müller, R.; Huber, C.G. Proteome Analysis of Myxococcus xanthus by Off-Line Two-Dimensional Chromatographic Separation Using Monolithic Poly-(styrene-divinylbenzene) Columns Combined with Ion-Trap Tandem Mass Spectrometry. J. Proteome Res. 2006, 5, 2760–2768. [Google Scholar] [CrossRef]
- Leinenbach, A.; Hartmer, R.; Lubeck, M.; Kneissl, B.; Elnakady, Y.A.; Baessmann, C.; Müller, R.; Huber, C.G. Proteome Analysis of Sorangium cellulosum Employing 2D-HPLC-MS/MS and Improved Database Searching Strategies for CID and ETD Fragment Spectra. J. Proteome Res. 2009, 8, 4350–4361. [Google Scholar] [CrossRef] [PubMed]
- Berleman, J.E.; Eallen, S.; Danielewicz, M.A.; Remis, J.P.; Egorur, A.; Ecunha, J.; Hadi, M.Z.; Zusman, D.R.; Northen, T.; Witkowska, H.E.; et al. The lethal cargo of Myxococcus xanthus outer membrane vesicles. Front. Microbiol. 2014, 5, 474. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, D.E.; Slade, S.E.; Mironas, A. Composition of distinct sub-proteomes in Myxococcus xanthus: Metabolic cost and amino acid availability. Amino Acids 2015, 47, 2521–2531. [Google Scholar] [CrossRef] [PubMed]
- Bolten, C.J.; Heinzle, E.; Müller, R.; Wittmann, C. Investigation of the central carbon metabolism of Sorangium cellulosum: Metabolic network reconstruction and quantification of pathway fluxes. J. Microbiol. Biotechnol. 2009, 19, 23–36. [Google Scholar] [PubMed]
- Robinson, M.; Son, B.; Kroos, D.; Kroos, L. Transcription factor MrpC binds to promoter regions of hundreds of developmentally-regulated genes in Myxococcus xanthus. BMC Genom. 2014, 15, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Mbp | %GC | Contigs | Released | Source | Accession |
|---|---|---|---|---|---|---|
| Anaeromyxobacter dehalogenans 2CP-C | 5.0 | 74.9 | 1 | Jan 2006 | [17] | GCA_000013385.1 |
| Myxococcus xanthus DK1622 | 9.1 | 68.9 | 1 | Jun 2006 | [18] | GCA_000012685.1 |
| Stigmatella aurantiaca DW4/3-1 | 10.3 | 67.4 | 579 | Sep 2006 | TIGR * | GCA_000168055.1 |
| Plesiocystis pacifica SIR-1T | 10.6 | 70.7 | 237 | Jun 2007 | G&BMF MGSP † | GCA_000170895.1 |
| Anaeromyxobacter sp. Fw109-5 | 5.3 | 73.5 | 1 | Jul 2007 | [28] | GCA_000017505.1 |
| Sorangium cellulosum So ce56 | 13.0 | 71.4 | 1 | Nov 2007 | [21] | GCA_000067165.1 |
| Anaeromyxobacter sp. K | 5.1 | 74.8 | 1 | Aug 2008 | US DOE JGI ‡ | GCA_000020805.1 |
| Anaeromyxobacter dehalogenans 2CP-1T | 5.0 | 74.7 | 1 | Jan 2009 | US DOE JGI ‡ | GCA_000022145.1 |
| Haliangium ochraceum SMP-2T | 9.5 | 69.5 | 1 | Oct 2009 | [23] | GCA_000024805.1 |
| Stigmatella aurantiaca DW4/3-1 | 10.3 | 67.5 | 1 | Oct 2010 | [24] | GCA_000165485.1 |
| Myxococcus macrosporus HW-1 | 9.0 | 70.6 | 1 | Jun 2011 | [26] | GCA_000219105.1 |
| Corallococcus coralloides DSM 2259T | 10.1 | 69.9 | 1 | Mar 2012 | [25] | GCA_000255295.1 |
| Strain | Taxonomy | Size (Mbp) | %GC | Contigs | CDS | L50 | N50 | Coverage | Genbank Accession | Area of Sampling | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CA046B | Corallococcus carmarthensis | 10.74 | 69.8 | 1079 | 8532 | 132 | 23389 | 71x | JABFJX000000000 | Carmarthen | [33] |
| CA044C | Corallococcus coralloides | 10.05 | 70 | 1379 | 8124 | 91 | 32261 | 65x | JABFJY000000000 | Carmarthen | [33] |
| AB043B | Corallococcus exercitus | 10.26 | 70.2 | 690 | 8102 | 119 | 26793 | 77x | JABFJV000000000 | Capel Bangor | [33] |
| CA046A | Corallococcus exercitus | 9.9 | 70.5 | 874 | 7710 | 146 | 21172 | 77x | JABFJW000000000 | Carmarthen | [33] |
| AB032A | Corallococcus exiguus | 10.44 | 69.6 | 972 | 8216 | 132 | 23729 | 60x | JABJTS000000000 | Penglais Woods, Aberystwyth | [33] |
| AB038A | Corallococcus exiguus | 10.57 | 69.4 | 631 | 8281 | 92 | 33924 | 72x | JABJTT000000000 | Penglais Woods, Aberystwyth | [33] |
| AB039A | Corallococcus exiguus | 10.54 | 69.4 | 716 | 8269 | 111 | 28467 | 91x | JABJTU000000000 | Penglais Woods, Aberystwyth | [33] |
| AM006 | Corallococcus exiguus | 10.59 | 69.5 | 865 | 8306 | 108 | 29172 | 67x | JABNNF000000000 | Gogerddan Farm, Aberystwyth | This study |
| AM007 | Corallococcus exiguus | 10.46 | 69.6 | 1264 | 8357 | 224 | 14471 | 29x | JABNNG000000000 | Gogerddan Farm, Aberystwyth | This study |
| CA046D | Corallococcus exiguus | 10.5 | 69.5 | 955 | 8310 | 122 | 26352 | 31x | JABNNE000000000 | Carmarthen | [33] |
| AM011 | Myxococcus eversor | 11.62 | 68.9 | 688 | 9104 | 110 | 33389 | 82x | JABXEM000000000 | Aberystwyth Harbour | This study |
| CA033 | Myxococcus llanfairensis * | 11.62 | 68.8 | 115 | 8928 | 9 | 312747 | 17x | JABUMU000000000 | Tanerdy Woods, Carmarthen | [33] |
| CA039A | Myxococcus llanfairensis * | 11.59 | 68.7 | 849 | 9039 | 133 | 27584 | 59x | JABUMQ000000000 | Tanerdy Woods, Carmarthen | [33] |
| CA040A | Myxococcus llanfairensis * | 11.72 | 68.9 | 70 | 8992 | 5 | 1036580 | 23x | JABUMR000000000 | Tanerdy Woods, Carmarthen | [33] |
| CA051A | Myxococcus llanfairensis * | 11.45 | 68.9 | 106 | 8765 | 7 | 562936 | 22x | JABUMS000000000 | Llansteffan | [33] |
| CA056 | Myxococcus llanfairensis * | 11.36 | 68.9 | 77 | 8742 | 5 | 658787 | 26x | JABUMT000000000 | Llansteffan | [33] |
| AM001 | Myxococcus vastator | 9.8 | 68.8 | 1946 | 8912 | 74 | 43403 | 115x | JABXEN000000000 | Anglesey | This study |
| AM009 | Myxococcus vastator | 8.8 | 70 | 550 | 6926 | 61 | 45904 | 138x | JABXEP000000000 | Clarach, Ceredigion | This study |
| AM010 | Myxococcus vastator | 8.93 | 70 | 276 | 6987 | 34 | 77358 | 125x | JABXEO000000000 | Gogerddan Farm, Aberystwyth | This study |
| AB023 | Myxococcus xanthus | 9.13 | 68.9 | 221 | 7181 | 30 | 102234 | 144x | JABFNQ000000000 | Gogerddan Farm, Aberystwyth | [33] |
| AM003 | Myxococcus xanthus | 9.14 | 69.2 | 380 | 7130 | 57 | 51156 | 109x | JABFNS000000000 | Anglesey | This study |
| AM005 | Myxococcus xanthus | 9.15 | 69.2 | 413 | 7148 | 53 | 55319 | 148x | JABFNT000000000 | Anglesey | This study |
| CA029 | Myxococcus xanthus | 9.19 | 68.8 | 584 | 7186 | 48 | 56679 | 93x | JABFNR000000000 | Carmarthen | [33] |
| CA059B | Pyxidicoccus fallax | 13.39 | 70.5 | 1321 | 10272 | 232 | 17466 | 25x | JABJTR000000000 | Llansteffan | [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitworth, D.E.; Sydney, N.; Radford, E.J. Myxobacterial Genomics and Post-Genomics: A Review of Genome Biology, Genome Sequences and Related ‘Omics Studies. Microorganisms 2021, 9, 2143. https://doi.org/10.3390/microorganisms9102143
Whitworth DE, Sydney N, Radford EJ. Myxobacterial Genomics and Post-Genomics: A Review of Genome Biology, Genome Sequences and Related ‘Omics Studies. Microorganisms. 2021; 9(10):2143. https://doi.org/10.3390/microorganisms9102143
Chicago/Turabian StyleWhitworth, David E., Natashia Sydney, and Emily J. Radford. 2021. "Myxobacterial Genomics and Post-Genomics: A Review of Genome Biology, Genome Sequences and Related ‘Omics Studies" Microorganisms 9, no. 10: 2143. https://doi.org/10.3390/microorganisms9102143
APA StyleWhitworth, D. E., Sydney, N., & Radford, E. J. (2021). Myxobacterial Genomics and Post-Genomics: A Review of Genome Biology, Genome Sequences and Related ‘Omics Studies. Microorganisms, 9(10), 2143. https://doi.org/10.3390/microorganisms9102143