
A Structural Approach to Anti-Virulence: A Discovery Pipeline


Abstract
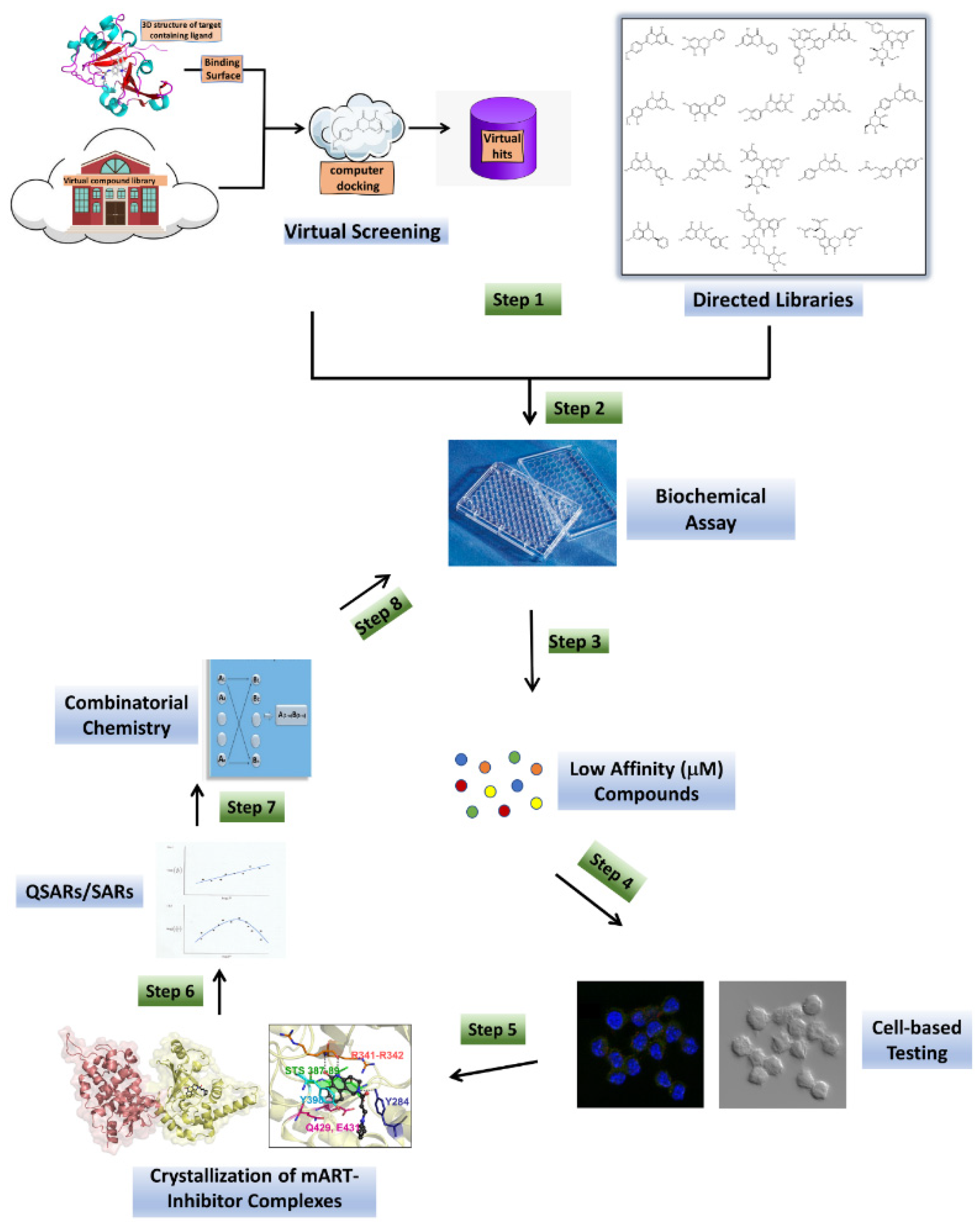
:1. Introduction
2. Materials and Methods
2.1. Recombinant Plx2A
2.2. Small Molecule Inhibitors of Plx2A
2.3. Glycohydrolase (GH) Activity
2.4. Inhibitor Binding to Plx2A
2.5. IC50 and Ki Determination
3. Results
3.1. Anti-Virulence Approach
3.1.1. Virtual Screens
3.1.2. Directed Libraries
3.1.3. Biochemical Assay
3.1.4. Cell-Based Testing
3.1.5. Crystallization of mART-Inhibitor Complexes
3.1.6. SARs/QSARs and Combinatorial Chemistry
3.2. Anti-Virulence Agents against American Foulbrood
3.2.1. Paenibacillus Larvae
3.2.2. C3larvin Toxin
3.2.3. Plx2A Toxin
4. Discussion
4.1. Flavonoids as Natural Product of Anti-Virulence Agents
4.2. Inhibition of C3-like mART Toxins
4.3. Anti-Virulence Strategy against American Foulbrood
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic Microbiol. 2021, 62, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Rubey, K.M.; Brenner, J.S. Nanomedicine to fight infectious disease. Adv. Drug Deliv. Rev. 2021, 179, 113996. [Google Scholar] [CrossRef]
- Tompson, A.C.; Manderson, L.; Chandler, C.I.R. Understanding antibiotic use: Practices, structures and networks. JAC Antimicrob. Resist. 2021, 3, dlab150. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.M.; Babakhani, S.; Moradi, L.; Karami, S.; Shahbandeh, M.; Mirshekar, M.; Mohebi, S.; Moghadam, M.T. Bacteriophage as a Novel Therapeutic Weapon for Killing Colistin-Resistant Multi-Drug-Resistant and Extensively Drug-Resistant Gram-Negative Bacteria. Curr. Microbiol. 2021, 78, 4023–4036. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.; Bhatia, S.; Singh, S.; Franco, F., Jr. Growing emergence of drug-resistant Pseudomonas aeruginosa and attenuation of its virulence using quorum sensing inhibitors: A critical review. Iran. J. Basic Med. Sci. 2021, 24, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zhang, L.; Kirienko, N.V. High-Throughput Approaches for the Identification of Pseudomonas aeruginosa Antivirulents. mBio 2021, 12, e02240-20. [Google Scholar] [CrossRef] [PubMed]
- Cherier, D.; Patin, D.; Blanot, D.; Touze, T.; Barreteau, H. The Biology of Colicin M and Its Orthologs. Antibiotics 2021, 10, 1109. [Google Scholar] [CrossRef] [PubMed]
- Hotinger, J.A.; Morris, S.T.; May, A.E. The Case against Antibiotics and for Anti-Virulence Therapeutics. Microorganisms 2021, 9, 2049. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, J.; Pieper, F.; Gobel, J.; Knispel, H.; McCarthy, M.; Goncalves, M.; Turner, M.; Merrill, A.R.; Genersch, E. Anti-Virulence Strategy against the Honey Bee Pathogenic Bacterium Paenibacillus larvae via Small Molecule Inhibitors of the Bacterial Toxin Plx2A. Toxins 2021, 13, 607. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.A.; Hurford, I.M.; Cassat, J.E. Antivirulence Strategies for the Treatment of Staphylococcus aureus Infections: A Mini Review. Front. Microbiol. 2020, 11, 632706. [Google Scholar] [CrossRef]
- Lim, K.Y.L.; Mullally, C.A.; Haese, E.C.; Kibble, E.A.; McCluskey, N.R.; Mikucki, E.C.; Thai, V.C.; Stubbs, K.A.; Sarkar-Tyson, M.; Kahler, C.M. Anti-Virulence Therapeutic Approaches for Neisseria gonorrhoeae. Antibiotics 2021, 10, 103. [Google Scholar] [CrossRef]
- Zhang, D.; Gan, R.Y.; Zhang, J.R.; Farha, A.K.; Li, H.B.; Zhu, F.; Wang, X.H.; Corke, H. Antivirulence properties and related mechanisms of spice essential oils: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1018–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.; Anwar, F.; Vedantam, G. Anti-virulence strategies for Clostridioides difficile infection: Advances and roadblocks. Gut Microbes 2020, 12, 1802865. [Google Scholar] [CrossRef] [PubMed]
- Yakovlieva, L.; Fulleborn, J.A.; Walvoort, M.T.C. Opportunities and Challenges of Bacterial Glycosylation for the Development of Novel Antibacterial Strategies. Front. Microbiol. 2021, 12, 745702. [Google Scholar] [CrossRef] [PubMed]
- Lakemeyer, M.; Zhao, W.; Mandl, F.A.; Hammann, P.; Sieber, S.A. Thinking outside the box—Novel antibacterials to tackle the resistance crisis. Angew. Chem. Int. Ed. 2018, 57, 14440–14475. [Google Scholar] [CrossRef] [PubMed]
- Lugo, M.R.; Merrill, A.R. Development of Anti-Virulence Therapeutics against Mono-ADP-Ribosyltransferase Toxins. Toxins 2020, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.P.; Molle, V. Staphylococcus aureus Toxins: An Update on Their Pathogenic Properties and Potential Treatments. Toxins 2021, 13, 677. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, M.; Sun, S. Pore-Forming Toxins During Bacterial Infection: Molecular Mechanisms and Potential Therapeutic Targets. Drug Des. Dev. Ther. 2021, 15, 3773–3781. [Google Scholar] [CrossRef]
- Kim, B.S. Spatiotemporal Regulation of Vibrio Exotoxins by HlyU and Other Transcriptional Regulators. Toxins 2020, 12, 544. [Google Scholar] [CrossRef]
- Vij, R.; Hube, B.; Brunke, S. Uncharted territories in the discovery of antifungal and antivirulence natural products from bacteria. Comput. Struct. Biotechnol. J. 2021, 19, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Hotinger, J.A.; Pendergrass, H.A.; May, A.E. Molecular Targets and Strategies for Inhibition of the Bacterial Type III Secretion System (T3SS); Inhibitors Directly Binding to T3SS Components. Biomolecules 2021, 11, 316. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Lyons, B.; Ravulapalli, R.; Lanoue, J.; Lugo, M.R.; Dutta, D.; Carlin, S.; Merrill, A.R. Scabin, a Novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 2016, 291, 11198–11215. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, G.; Corda, D.; Catara, G. From toxins to mammalian enzymes: The diversity of mono-ADP-ribosylation. Front. Biosci. Landmark Ed. 2015, 20, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Tsuge, H. Common Mechanism for Target Specificity of Protein- and DNA-Targeting ADP-Ribosyltransferases. Toxins 2021, 13, 40. [Google Scholar] [CrossRef]
- Aktories, K.; Lang, A.E.; Schwan, C.; Mannherz, H.G. Actin as target for modification by bacterial protein toxins. FEBS J. 2011, 278, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Vogelsgesang, M.; Pautsch, A.; Aktories, K. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedeberg’s Arch. Pharmacol. 2007, 374, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Fieldhouse, R.J.; Turgeon, Z.; White, D.; Merrill, A.R. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 2010, 6, e1001029. [Google Scholar] [CrossRef] [PubMed]
- Fieldhouse, R.J.; Merrill, A.R. Needle in the haystack: Structure-based toxin discovery. Trends Biochem. Sci. 2008, 33, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, O.; Thow, Z.; Geddes-McAlister, J.; Merrill, A.R. Several New Putative Bacterial ADP-Ribosyltransferase Toxins Are Revealed from In Silico Data Mining, Including the Novel Toxin Vorin, Encoded by the Fire Blight Pathogen, Erwinia amylovora. Toxins 2020, 12, 792. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Soding, J.; Remmert, M.; Biegert, A.; Lupas, A.N. HHsenser: Exhaustive transitive profile search using HMM-HMM comparison. Nucleic Acids Res. 2006, 34, W374–W378. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, Z.; White, D.; Jorgensen, R.; Visschedyk, D.; Fieldhouse, R.J.; Mangroo, D.; Merrill, A.R. Yeast as a tool for characterizing mono-ADP-ribosyltransferase toxins. FEMS Microbiol. Lett. 2009, 300, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.; Li, J.H.; Zhang, J.; Merrill, A.R. Characterization of competitive inhibitors for the transferase activity of Pseudomonas aeruginosa exotoxin A. J. Enzym. Inhib. Med. Chem. 2002, 17, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Yates, S.P.; Taylor, P.L.; Jorgensen, R.; Ferraris, D.; Zhang, J.; Andersen, G.R.; Merrill, A.R. Structure-function analysis of water-soluble inhibitors of the catalytic domain of exotoxin A from Pseudomonas aeruginosa. Biochem. J. 2005, 385, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, R.; Merrill, A.R.; Yates, S.P.; Marquez, V.E.; Schwan, A.L.; Boesen, T.; Andersen, G.R. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 2005, 436, 979–984. [Google Scholar] [CrossRef]
- Thompson, C.; Merrill, A.R.; Mangroo, D. Identification of peptide inhibitors of Pseudomonas aeruginosa exotoxin A function using a yeast two-hybrid approach. FEMS Microbiol. Lett. 2003, 218, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.; Merrill, A.R. Toward the elucidation of the catalytic mechanism of the mono-ADP-ribosyltransferase activity of Pseudomonas aeruginosa exotoxin A. Biochemistry 2004, 43, 183–194. [Google Scholar] [CrossRef]
- Beattie, B.K.; Prentice, G.A.; Merrill, A.R. Investigation into the catalytic role for the tryptophan residues within domain III of Pseudomonas aeruginosa exotoxin A. Biochemistry 1996, 35, 15134–15142. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Lajoie, G.; Merrill, A.R. The role of the diphthamide-containing loop within eukaryotic elongation factor 2 in ADP-ribosylation by Pseudomonas aeruginosa exotoxin A. Biochem. J. 2008, 413, 163–174. [Google Scholar] [CrossRef]
- Yates, S.P.; Merrill, A.R. Elucidation of eukaryotic elongation factor-2 contact sites within the catalytic domain of Pseudomonas aeruginosa exotoxin A. Biochem. J. 2004, 379, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, F.; Prentice, G.A.; Merrill, A.R. Protein-protein interaction using tryptophan analogues: Novel spectroscopic probes for toxin-elongation factor-2 interactions. Biochemistry 2001, 40, 10273–10283. [Google Scholar] [CrossRef]
- Yates, S.P.; Merrill, A.R. A catalytic loop within Pseudomonas aeruginosa exotoxin A modulates its transferase activity. J. Biol. Chem. 2001, 276, 35029–35036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.; Merrill, A.R. Application of a fluorometric assay for characterization of the catalytic competency of a domain III fragment of Pseudomonas aeruginosa exotoxin A. Anal. Biochem. 2001, 292, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Beattie, B.K.; Merrill, A.R. A fluorescence investigation of the active site of Pseudomonas aeruginosa exotoxin A. J. Biol. Chem. 1999, 274, 15646–15654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, B.K.; Merrill, A.R. In vitro enzyme activation and folded stability of Pseudomonas aeruginosa exotoxin A and its C-terminal peptide. Biochemistry 1996, 35, 9042–9051. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, Z.; Jorgensen, R.; Visschedyk, D.; Edwards, P.R.; Legree, S.; McGregor, C.; Fieldhouse, R.J.; Mangroo, D.; Schapira, M.; Merrill, A.R. Newly discovered and characterized antivirulence compounds inhibit bacterial mono-ADP-ribosyltransferase toxins. Antimicrob. Agents Chemother. 2011, 55, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugo, M.R.; Ravulapalli, R.; Dutta, D.; Merrill, A.R. Structural variability of C3larvin toxin. Intrinsic dynamics of the α/β fold of the C3-like group of mono-ADP-ribosyltransferase toxins. J. Biomol. Struct. Dyn. 2016, 34, 2537–2560. [Google Scholar] [CrossRef]
- Lugo, M.R.; Merrill, A.R. Pocket analysis of the full-length cholix toxin. An assessment of the structure-dynamics of the apo catalytic domain. J. Biomol. Struct. Dyn. 2015, 33, 2452–2468. [Google Scholar] [CrossRef]
- Lugo, M.R.; Merrill, A.R. The Father, Son and Cholix Toxin: The Third Member of the DT Group Mono-ADP-Ribosyltransferase Toxin Family. Toxins 2015, 7, 2757–2772. [Google Scholar] [CrossRef] [Green Version]
- Lugo, M.R.; Merrill, A.R. A comparative structure-function analysis of active-site inhibitors of Vibrio cholerae cholix toxin. J. Mol. Recognit. 2015, 28, 539–552. [Google Scholar] [CrossRef]
- Fieldhouse, R.J.; Jorgensen, R.; Lugo, M.R.; Merrill, A.R. The 1.8 Å cholix toxin crystal structure in complex with NAD+ and evidence for a new kinetic model. J. Biol. Chem. 2012, 287, 21176–21188. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, R.; Wang, Y.; Visschedyk, D.; Merrill, A.R. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 2008, 9, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, R.; Purdy, A.E.; Fieldhouse, R.J.; Kimber, M.S.; Bartlett, D.H.; Merrill, A.R. Cholix Toxin, a Novel ADP-ribosylating Factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visschedyk, D.; Rochon, A.; Tempel, W.; Dimov, S.; Park, H.W.; Merrill, A.R. Certhrax toxin, an anthrax-related ADP-ribosyltransferase from Bacillus cereus. J. Biol. Chem. 2012, 287, 41089–41102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visschedyk, D.D.; Perieteanu, A.A.; Turgeon, Z.J.; Fieldhouse, R.J.; Dawson, J.F.; Merrill, A.R. Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J. Biol. Chem. 2010, 285, 13525–13534. [Google Scholar] [CrossRef] [Green Version]
- Perieteanu, A.A.; Visschedyk, D.D.; Merrill, A.R.; Dawson, J.F. ADP-ribosylation of cross-linked actin generates barbed-end polymerization-deficient F-actin oligomers. Biochemistry 2010, 49, 8944–8954. [Google Scholar] [CrossRef] [PubMed]
- Shniffer, A.; Visschedyk, D.D.; Ravulapalli, R.; Suarez, G.; Turgeon, Z.J.; Petrie, A.A.; Chopra, A.K.; Merrill, A.R. Characterization of an actin-targeting ADP-ribosyltransferase from Aeromonas hydrophila. J. Biol. Chem. 2012, 287, 37030–37041. [Google Scholar] [CrossRef] [Green Version]
- Krska, D.; Ravulapalli, R.; Fieldhouse, R.J.; Lugo, M.R.; Merrill, A.R. C3larvin Toxin, an ADP-ribosyltransferase from Paenibacillus larvae. J. Biol. Chem. 2015, 290, 1639–1653. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, J.; Funfhaus, A.; Knispel, H.; Krska, D.; Ravulapalli, R.; Heney, K.A.; Lugo, M.R.; Merrill, A.R.; Genersch, E. Characterization of the toxin Plx2A, a RhoA-targeting ADP-ribosyltransferase produced by the honey bee pathogen Paenibacillus larvae. Environ. Microbiol. 2017, 19, 5100–5116. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Tremblay, O.; Heney, K.A.; Lugo, M.R.; Ebeling, J.; Genersch, E.; Merrill, A.R. Characterization of C3larvinA, a novel RhoA-targeting ADP-ribosyltransferase toxin produced by the honey bee pathogen, Paenibacillus larvae. Biosci. Rep. 2020, 40, BSR20193405. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Heney, K.A.; Merrill, A.R. The N-terminus of Paenibacillus larvae C3larvinA modulates catalytic efficiency. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef] [PubMed]
- Ravulapalli, R.; Lugo, M.R.; Pfoh, R.; Visschedyk, D.; Poole, A.; Fieldhouse, R.J.; Pai, E.F.; Merrill, A.R. Characterization of Vis Toxin, a Novel ADP-Ribosyltransferase from Vibrio splendidus. Biochemistry 2015, 54, 5920–5936. [Google Scholar] [CrossRef] [PubMed]
- Lyons, B.; Lugo, M.R.; Carlin, S.; Lidster, T.; Merrill, A.R. Characterization of the catalytic signature of Scabin toxin, a DNA-targeting ADP-ribosyltransferase. Biochem. J. 2018, 475, 225–245. [Google Scholar] [CrossRef]
- Lugo, M.R.; Lyons, B.L.; Lento, C.; Wilson, D.J.; Merrill, A.R. Dynamics of Scabin toxin. A proposal for the binding mode of the DNA substrate. PLoS ONE 2018, 13, e0194425. [Google Scholar] [CrossRef]
- Vatta, M.; Lyons, B.; Heney, K.A.; Lidster, T.; Merrill, A.R. Mapping the DNA-Binding Motif of Scabin Toxin, a Guanine Modifying Enzyme from Streptomyces scabies. Toxins 2021, 13, 55. [Google Scholar] [CrossRef]
- Swann, S.L.; Brown, S.P.; Muchmore, S.W.; Patel, H.; Merta, P.; Locklear, J.; Hajduk, P.J. A unified, probabilistic framework for structure- and ligand-based virtual screening. J. Med. Chem. 2011, 54, 1223–1232. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef]
- Rollinger, J.M.; Stuppner, H.; Langer, T. Virtual screening for the discovery of bioactive natural products. Prog. Drug Res. 2008, 65, 211, 213–249. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, N.M.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Virtual screening of compound libraries. Methods Mol. Biol. 2009, 572, 57–70. [Google Scholar] [PubMed]
- Sousa, S.F.; Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Virtual screening in drug design and development. Comb. Chem. High Throughput Screen. 2010, 13, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Raushel, F.M.; Shoichet, B.K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 2005, 44, 12316–12328. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin, M.; Khan, A.U. Structure based virtual screening to discover putative drug candidates: Necessary considerations and successful case studies. Methods 2015, 71, 135–145. [Google Scholar] [CrossRef]
- Elseginy, S.A. Virtual screening and structure-based 3D pharmacophore approach to identify small-molecule inhibitors of SARS-CoV-2 Mpro. J. Biomol. Struct. Dyn. 2021, 1–17. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Babii, C.; Mihalache, G.; Bahrin, L.G.; Neagu, A.N.; Gostin, I.; Mihai, C.T.; Sarbu, L.G.; Birsa, L.M.; Stefan, M. A novel synthetic flavonoid with potent antibacterial properties: In vitro activity and proposed mode of action. PLoS ONE 2018, 13, e0194898. [Google Scholar] [CrossRef] [Green Version]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure-activity relationship: An update review. Phytother. Res. 2019, 33, 13–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Khulbe, M.; Aggarwal, A.; Kathuria, A. Recent advances in continuous flow synthesis of heterocycles. Mol. Divers. 2021. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Hamashima, Y. Practical and Scalable Organic Reactions with Flow Microwave Apparatus. Chem. Rec. 2019, 19, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, M.A.; Zhang, W.P.; Mallik, D.; Organ, M.G. Sampling and Analysis in Flow: The Keys to Smarter, More Controllable, and Sustainable Fine-Chemical Manufacturing. Angew. Chem. Int. Ed. 2021, 60, 20606–20626. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Elshafiey, E.H.; Shetaia, A.A.; El-Wahed, A.A.A.; Algethami, A.F.; Musharraf, S.G.; AlAjmi, M.F.; Zhao, C.; Masry, S.H.D.; Abdel-Daim, M.M.; et al. Overview of Bee Pollination and Its Economic Value for Crop Production. Insects 2021, 12, 688. [Google Scholar] [CrossRef] [PubMed]
- Poppinga, L.; Genersch, E. Molecular pathogenesis of American Foulbrood: How Paenibacillus larvae kills honey bee larvae. Curr. Opin. Insect Sci. 2015, 10, 29–36. [Google Scholar] [CrossRef]
- Applegate, J.R., Jr.; Petritz, O.A. Common and Emerging Infectious Diseases of Honeybees (Apis mellifera). Vet. Clin. Exot. Anim. Pract. 2020, 23, 285–297. [Google Scholar] [CrossRef]
- Ebeling, J.; Knispel, H.; Hertlein, G.; Funfhaus, A.; Genersch, E. Biology of Paenibacillus larvae, a deadly pathogen of honey bee larvae. Appl. Microbiol. Biotechnol. 2016, 100, 7387–7395. [Google Scholar] [CrossRef]
- Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 2010, 103 (Suppl. 1), S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Beims, H.; Bunk, B.; Erler, S.; Mohr, K.I.; Sproer, C.; Pradella, S.; Gunther, G.; Rohde, M.; von der Ohe, W.; Steinert, M. Discovery of Paenibacillus larvae ERIC V: Phenotypic and genomic comparison to genotypes ERIC I–IV reveal different inventories of virulence factors which correlate with epidemiological prevalences of American Foulbrood. Int. J. Med. Microbiol. 2020, 310, 151394. [Google Scholar] [CrossRef]
- Djukic, M.; Brzuszkiewicz, E.; Funfhaus, A.; Voss, J.; Gollnow, K.; Poppinga, L.; Liesegang, H.; Garcia-Gonzalez, E.; Genersch, E.; Daniel, R. How to kill the honey bee larva: Genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS ONE 2014, 9, e90914. [Google Scholar] [CrossRef] [PubMed]
- Funfhaus, A.; Poppinga, L.; Genersch, E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 2013, 15, 2951–2965. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, J.; Funfhaus, A.; Genersch, E. The Buzz about ADP-Ribosylation Toxins from Paenibacillus larvae, the Causative Agent of American Foulbrood in Honey Bees. Toxins 2021, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, X.; Wang, Z.; Liu, Y.; Guo, J.; Zhu, Y.; Shao, J.; Li, J.; Wang, L.; Wang, K. Antibacterial, antioxidant and biocompatible nanosized quercetin-PVA xerogel films for wound dressing. Colloids Surf. B Biointerfaces 2021, 209, 112175. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Chemical Name | Structure | a KD (μmol/L) | b IC50 (μmol/L) |
|---|---|---|---|---|
| Acacetin | 5,7-dihydroxy-2-(4-methoxy phenyl)-4H-chromen-4-one |  | c ND | 28.0 ± 2.3 |
| Baicalein | 5,6,7-trihydroxy-2-phenyl-4H-chromen-4-one |  | 13.6 ± 1.5 | 10.7 ± 0.7 |
| Chrysin | 5,7-dihydroxy-2-phenyl-4H-chromen-4-one |  | 40.6 ± 4.7 | 49.2 ± 2.5 |
| Jaceosidin | 5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-6-methoxy-4H-chromen-4-one |  | 24.1 ± 2.9 | 73.9 ± 4.7 |
| Kaempferol | 3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one |  | 37.3 ± 3.6 | 79.9 ± 6.0 |
| Luteolin | 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one |  | 24.0 ± 1.5 | 68.1 ± 1.8 |
| Morin | 2-(2,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one |  | 32.8 ± 11.7 | 109 ± 4.8 |
| Quercetin | 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one |  | 19.9 ± 0.4 | 24.3 ± 0.8 |
| M3 | N-{[(3R)-1-{1H-pyrazolo[3,4-d]pyrimidin-4- yl}piperidin-3-yl]methyl}methanesulfon amide |  | c ND | 216.3 ± 22.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarthy, M.; Goncalves, M.; Powell, H.; Morey, B.; Turner, M.; Merrill, A.R. A Structural Approach to Anti-Virulence: A Discovery Pipeline. Microorganisms 2021, 9, 2514. https://doi.org/10.3390/microorganisms9122514
McCarthy M, Goncalves M, Powell H, Morey B, Turner M, Merrill AR. A Structural Approach to Anti-Virulence: A Discovery Pipeline. Microorganisms. 2021; 9(12):2514. https://doi.org/10.3390/microorganisms9122514
Chicago/Turabian StyleMcCarthy, Michael, Monica Goncalves, Hannah Powell, Blake Morey, Madison Turner, and Allan Rod Merrill. 2021. "A Structural Approach to Anti-Virulence: A Discovery Pipeline" Microorganisms 9, no. 12: 2514. https://doi.org/10.3390/microorganisms9122514
APA StyleMcCarthy, M., Goncalves, M., Powell, H., Morey, B., Turner, M., & Merrill, A. R. (2021). A Structural Approach to Anti-Virulence: A Discovery Pipeline. Microorganisms, 9(12), 2514. https://doi.org/10.3390/microorganisms9122514