
The Roles of the Virome in Cancer



Abstract
:1. Introduction—The Human Virome
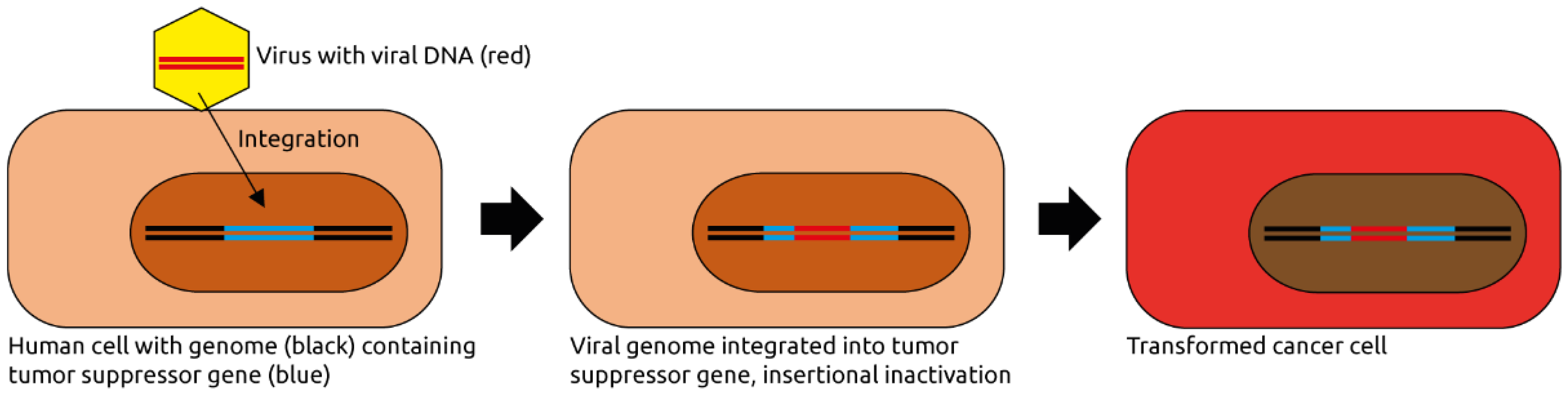
2. The Roles of the Eukaryotic Virome in Cancer
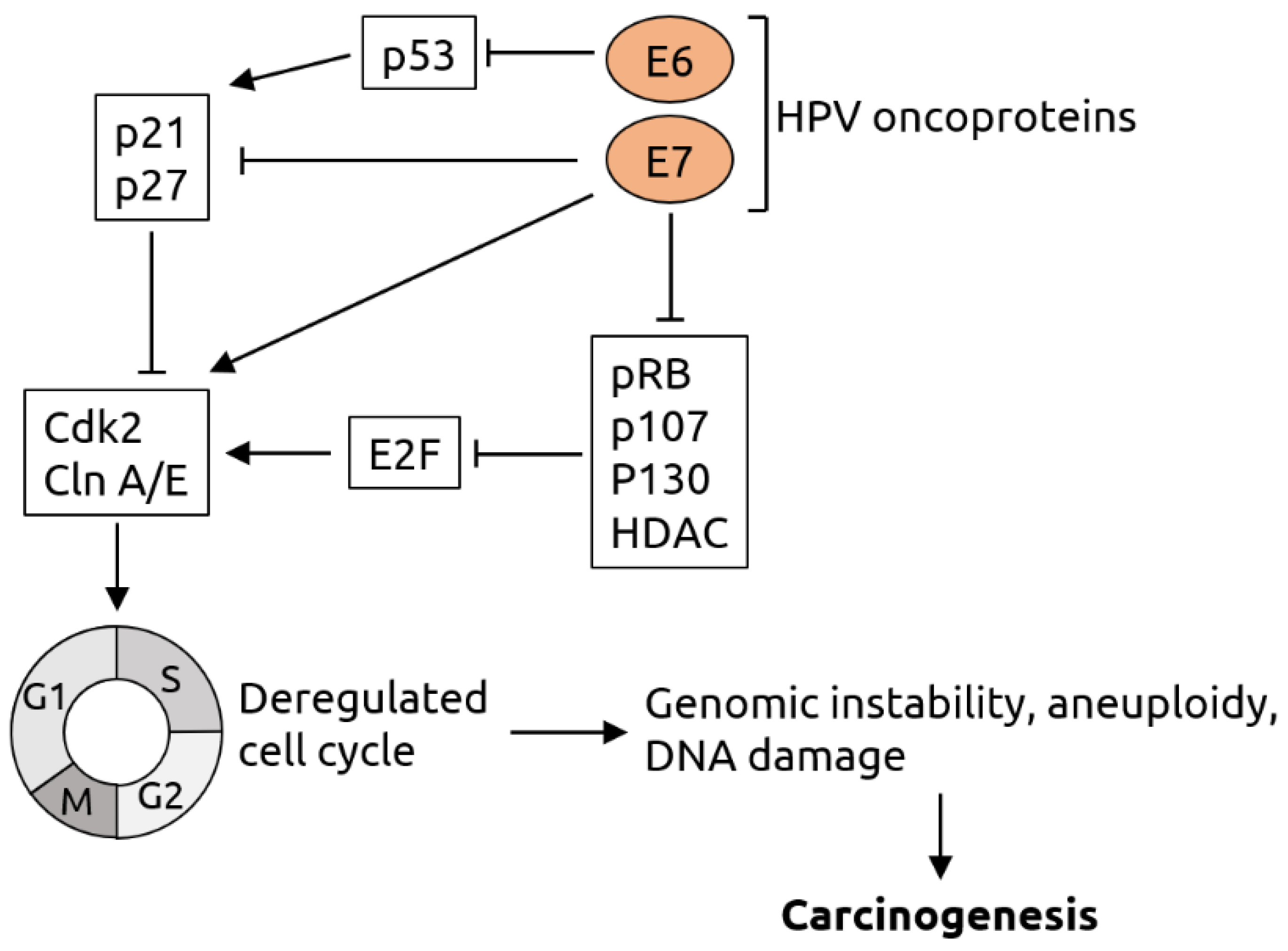
2.1. Papillomaviridae
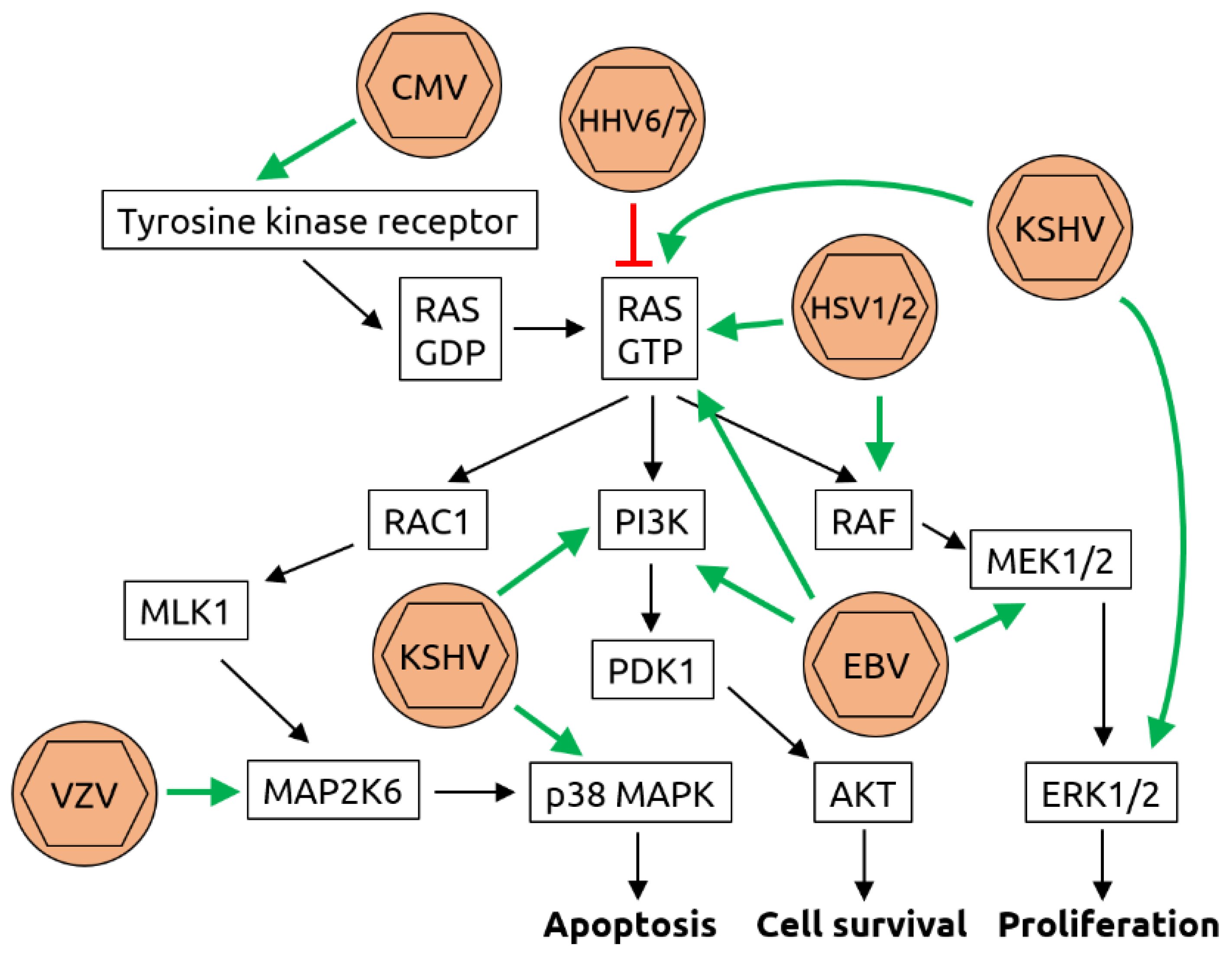
2.2. Herpesviridae
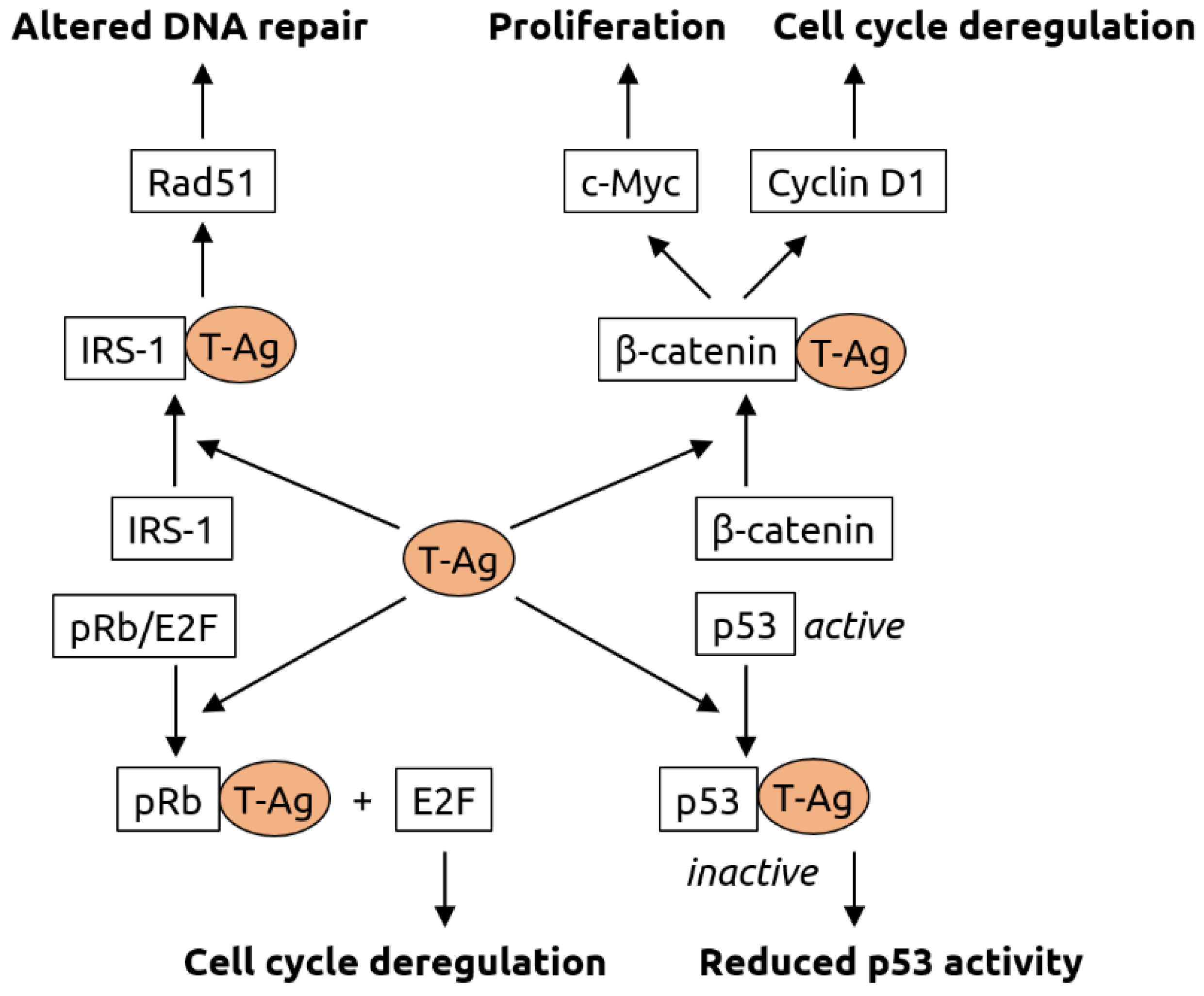
2.3. Polyomaviridae
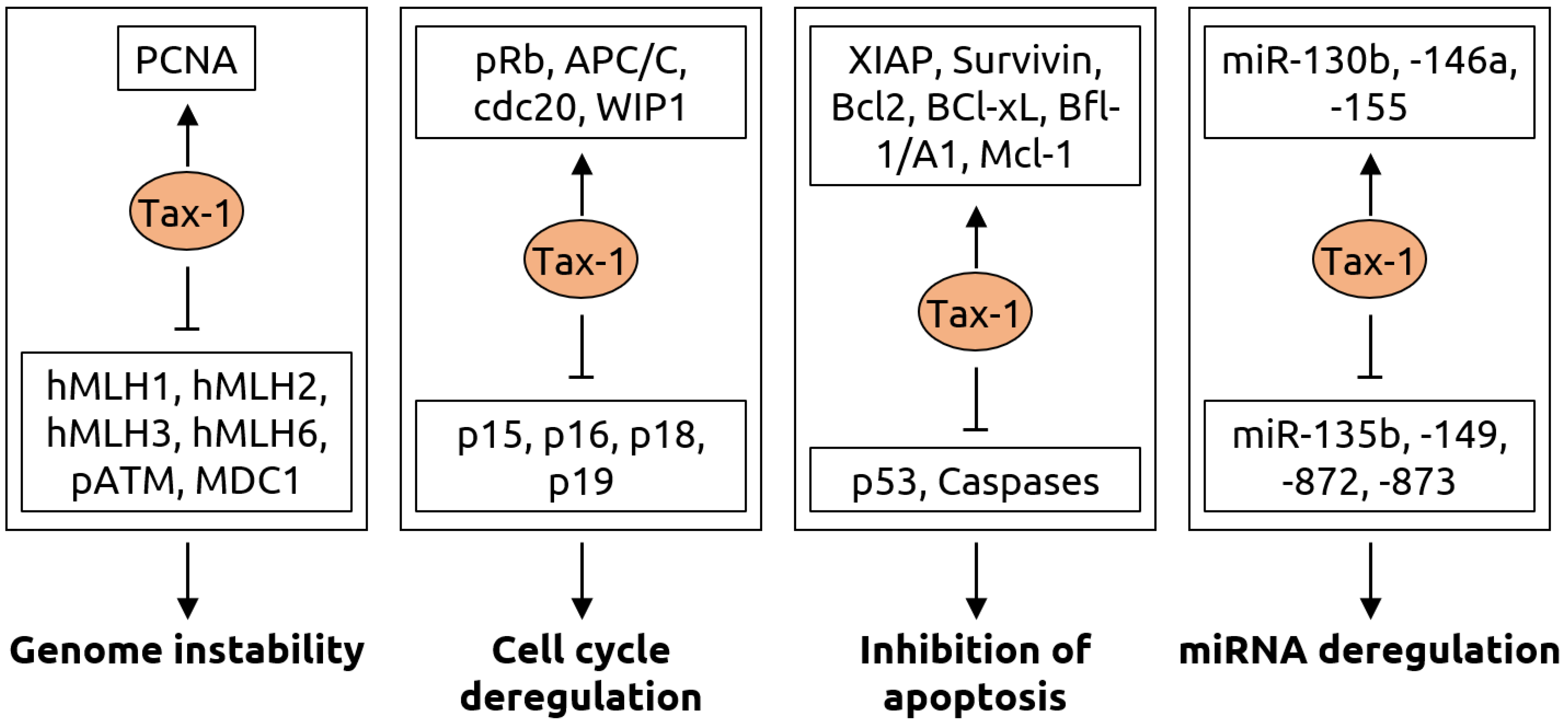
2.4. Retroviridae
2.5. Other Viruses
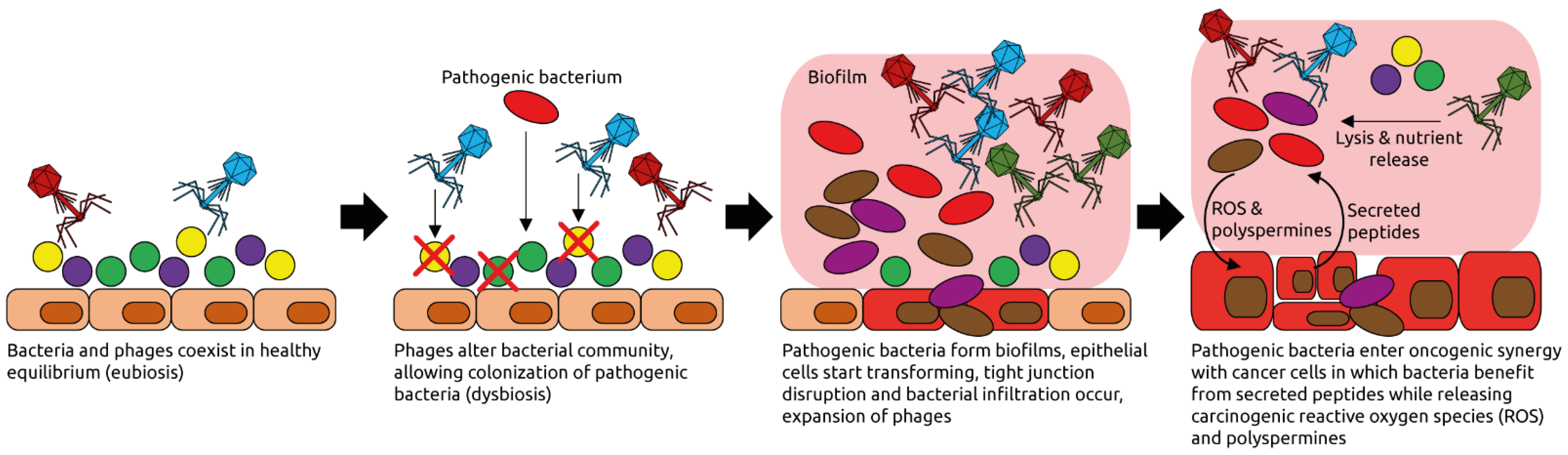
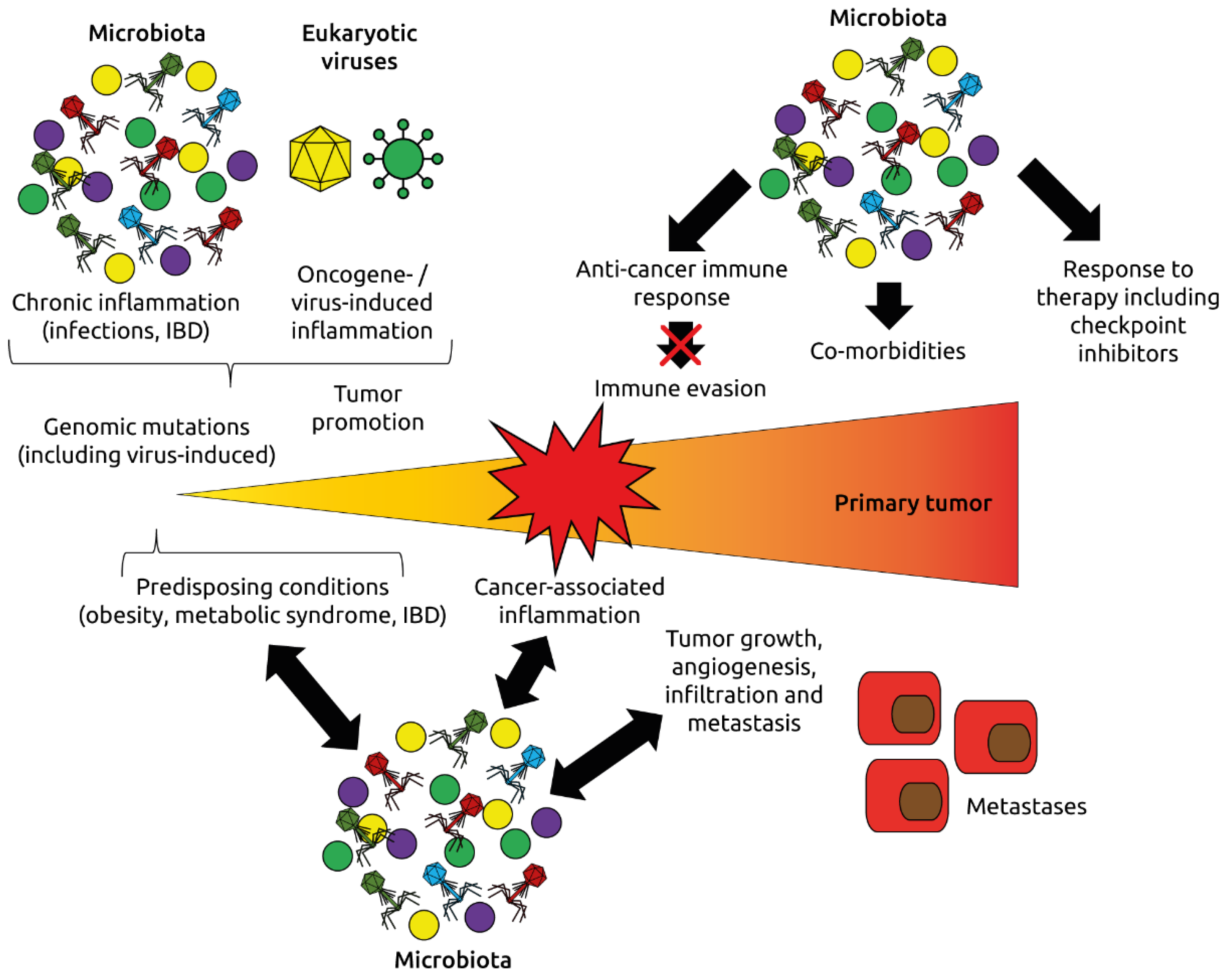
3. The Human Intestinal Virome and Its Links to Cancer
4. Fecal Microbiota Transplantation—Focus on Viruses and Cancer
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown“ of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.L.; Wie, J.Y.; Wang, L.; Huang, S.L.; Chen, J.L. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharmacol. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marônek, M.; Link, R.; Monteleone, G.; Gardlík, R.; Stolfi, C. Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders? Int. J. Mol. Sci. 2020, 21, 8133. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, S.; Yamanegi, K.; Xiao, S.Y.; Da Costa, M.; Palefsky, J.M.; Zheng, Z.M. Colorectal papillomavirus infection in patients with colorectal cancer. Clin. Cancer Res. 2005, 11, 2862–2867. [Google Scholar] [CrossRef] [Green Version]
- Muresu, N.; Sotgiu, G.; Saderi, L.; Sechi, I.; Cossu, A.; Marras, V.; Meloni, M.; Martinelli, M.; Cocuzza, C.; Tanda, F.; et al. Distribution of HPV Genotypes in Patients with a Diagnosis of Anal Cancer in an Italian Region. Int. J. Environ. Res. Public Health 2020, 17, 4516. [Google Scholar] [CrossRef]
- Kabarriti, R.; Brodin, N.P.; Ohri, N.; Narang, R.; Huang, R.; Chuy, J.W.; Rajdev, L.N.; Kalnicki, S.; Guha, C.; Garg, M.K. Human papillomavirus, radiation dose and survival of patients with anal cancer. Acta Oncol. 2019, 58, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.G.; Katz, J.P.; Pipas, J.M. Viral sequences in human cancer. Virology 2018, 513, 208–216. [Google Scholar] [CrossRef] [PubMed]
- de Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Clifford, G.; Franceschi, S.; Diaz, M.; Muñoz, N.; Villa, L.L. Chapter 3: HPV type-distribution in women with and without cervical neoplastic diseases. Vaccine 2006, 24 (Suppl. S3), 26–34. [Google Scholar] [CrossRef]
- Kirgan, D.; Manalo, P.; Hall, M.; McGregor, B. Association of human papillomavirus and colon neoplasms. Arch. Surg. 1990, 125, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Leu, S.Y.; Chiang, H.; Fung, C.P.; Liu, W.T. Human papillomavirus type 18 in colorectal cancer. J. Microbiol. Immunol. Infect. 2001, 34, 87–91. [Google Scholar] [PubMed]
- Fiorina, L.; Ricotti, M.; Vanoli, A.; Luinetti, O.; Dallera, E.; Riboni, R.; Paolucci, S.; Brugnatelli, S.; Paulli, M.; Pedrazzoli, P.; et al. Systematic analysis of human oncogenic viruses in colon cancer revealed EBV latency in lymphoid infiltrates. Infect. Agent. Cancer 2014, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Bjørge, T.; Hakulinen, T.; Engeland, A.; Jellum, E.; Koskela, P.; Lehtinen, M.; Luostarinen, T.; Paavonen, J.; Sapp, M.; Schiller, J.; et al. A prospective, seroepidemiological study of the role of human papillomavirus in esophageal cancer in Norway. Cancer Res. 1997, 57, 3989–3992. [Google Scholar] [PubMed]
- Zhang, S.K.; Guo, L.W.; Chen, Q.; Zhang, M.; Liu, S.Z.; Quan, P.L.; Lu, J.B.; Sun, X.B. The association between human papillomavirus 16 and esophageal cancer in Chinese population: A meta-analysis. BMC Cancer 2015, 15, 1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tian, X.; Liu, F.; Zhao, Y.; Sun, M.; Chen, D.; Lu, C.; Wang, Z.; Shi, X.; Zhang, Q.; et al. Detection of HPV DNA in esophageal cancer specimens from different regions and ethnic groups: A descriptive study. BMC Cancer 2010, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; TCGA Network; Meric-Bernstam, F.; Medeiros, L.J.; et al. Landscape of DNA virus associations across human malignant cancers: Analysis of 3775 cases using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [Green Version]
- Curado, M.P.; Hashibe, M. Recent changes in the epidemiology of head and neck cancer. Curr. Opin. Oncol. 2009, 21, 194–200. [Google Scholar] [CrossRef]
- Maden, C.; Beckmann, A.M.; Thomas, D.B.; McKnight, B.; Sherman, K.J.; Ashley, R.L.; Corey, L.; Daling, J.R. Human papillomaviruses, herpes simplex viruses, and the risk of oral cancer in men. Am. J. Epidemiol. 1992, 135, 1093–1102. [Google Scholar] [CrossRef]
- Bae, J.M. Human papillomavirus 16 infection as a potential risk factor for prostate cancer: An adaptive meta-analysis. Epidemiol. Health 2015, 37, e2015005. [Google Scholar] [CrossRef] [Green Version]
- Farhadi, A.; Behzad-Behbahani, A.; Geramizadeh, B.; Sekawi, Z.; Rahsaz, M.; Sharifzadeh, S. High-risk human papillomavirus infection in different histological subtypes of renal cell carcinoma. J. Med. Virol. 2014, 86, 1134–1144. [Google Scholar] [CrossRef]
- Mollerup, S.; Asplund, M.; Friis-Nielsen, J.; Kjartansdóttir, K.R.; Fridholm, H.; Hansen, T.A.; Herrera, J.A.R.; Barnes, C.J.; Jensen, R.H.; Richter, S.R.; et al. High-Throughput Sequencing-Based Investigation of Viruses in Human Cancers by Multienrichment Approach. J. Infect. Dis. 2019, 220, 1312–1324. [Google Scholar] [CrossRef]
- Gewirtzman, A.; Bartlett, B.; Tyring, S. Epidermodysplasia verruciformis and human papilloma virus. Curr. Opin. Infect. Dis. 2008, 21, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ramoz, N.; Rueda, L.A.; Bouadjar, B.; Montoya, L.S.; Orth, G.; Favre, M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 2002, 32, 579–581. [Google Scholar] [CrossRef]
- Rakislova, N.; Saco, A.; Sierra, A.; Del Pino, M.; Ordi, J. Role of Human Papillomavirus in Vulvar Cancer. Adv. Anat. Pathol. 2017, 24, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, J.; Hong, T.T.; Skarstedt, M.; Löfgren, S.; Zar, N.; Matussek, A. Detection of cytomegalovirus DNA in colorectal tissue from Swedish and Vietnamese patients with colorectal cancer. Anticancer Res. 2013, 33, 4947–4950. [Google Scholar]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Yang, C.Y.; Chen, Y.C.; Lin, C.H.; Chou, T.Y.; Cho, W.L.; Chan, Y.J. Identification of human cytomegalovirus in tumour tissues of colorectal cancer and its association with the outcome of non-elderly patients. J. Gen. Virol. 2016, 97, 2411–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.P.; Jiang, J.K.; Chan, C.H.; Teo, W.H.; Yang, C.Y.; Chen, Y.C.; Chou, T.Y.; Lin, C.H.; Chan, Y.J. Genetic polymorphisms of the human cytomegalovirus UL144 gene in colorectal cancer and its association with clinical outcome. J. Gen. Virol. 2015, 96, 3613–3623. [Google Scholar] [CrossRef]
- Song, L.B.; Zhang, X.; Zhang, C.Q.; Zhang, Y.; Pan, Z.Z.; Liao, W.T.; Li, M.Z.; Zeng, M.S. Infection of Epstein-Barr virus in colorectal cancer in Chinese. Ai Zheng 2006, 25, 1356–1360. [Google Scholar]
- Awerkiew, S.; Bollschweiler, E.; Metzger, R.; Schneider, P.M.; Hölscher, A.H.; Pfister, H. Esophageal cancer in Germany is associated with Epstein-Barr-virus but not with papillomaviruses. Med. Microbiol. Immunol. 2003, 192, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, J.L.; Torres, J.; Camorlinga-Ponce, M.; Mantilla, A.; Leal, Y.A.; Fuentes-Pananá, E.M. Evidence of Epstein-Barr virus association with gastric cancer and non-atrophic gastritis. Viruses 2014, 6, 301–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corallo, S.; Fucà, G.; Morano, F.; Salati, M.; Spallanzani, A.; Gloghini, A.; Volpi, C.C.; Trupia, D.V.; Lobefaro, R.; Guarini, V.; et al. Clinical Behavior and Treatment Response of Epstein-Barr Virus-Positive Metastatic Gastric Cancer: Implications for the Development of Future Trials. Oncologist 2020, 25, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, B.A.; Zeng, Y.M.; Chen, G.C.; Li, X.X.; Chen, J.T.; Guo, Y.W.; Li, M.H.; Zeng, Y. Epstein-Barr virus in hepatocellular carcinogenesis. World J. Gastroenterol. 2004, 10, 3409–3413. [Google Scholar] [CrossRef]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein-Barr virus and Burkitt lymphoma. J. Clin. Pathol. 2007, 60, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; O’Grady, T.; Lin, Z.; Xu, G.; Baddoo, M.; Parsons, C.; Zhang, K.; Taylor, C.M.; Flemington, E.K. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin’s diffuse large B-cell lymphoma cohort by using RNA sequencing. J. Virol. 2013, 87, 13059–13062. [Google Scholar] [CrossRef] [Green Version]
- Nakhoul, H.; Lin, Z.; Wang, X.; Roberts, C.; Dong, Y.; Flemington, E. High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments. mSphere 2019, 4, e00248-19. [Google Scholar] [CrossRef] [Green Version]
- Jalouli, J.; Jalouli, M.M.; Sapkota, D.; Ibrahim, S.O.; Larsson, P.A.; Sand, L. Human papilloma virus, herpes simplex virus and epstein barr virus in oral squamous cell carcinoma from eight different countries. Anticancer Res. 2012, 32, 571–580. [Google Scholar]
- Goncalves, P.H.; Montezuma-Rusca, J.M.; Yarchoan, R.; Uldrick, T.S. Cancer prevention in HIV-infected populations. Semin. Oncol. 2016, 43, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, M.; Szymanski, J.; Slavcheva, E.; Rao, A.; Kelly, A.; Jones, K.; Jaffers, G. BK virus associated renal cell carcinoma: Case presentation with optimized PCR and other diagnostic tests. Am. J. Transplant. 2007, 7, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Jiang, M.; Imperiale, M.J. BK virus and human cancer: Innocent until proven guilty. Semin. Cancer Biol. 2009, 19, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Shoraka, H.R.; Abobakri, O.; Naghibzade Tahami, A.; Mollaei, H.R.; Bagherinajad, Z.; Malekpour Afshar, R.; Shahesmaeili, A. Prevalence of JC and BK viruses in Patients with Colorectal Cancer: A Systematic Review and Meta- Analysis. Asian Pac. J. Cancer Prev. 2020, 21, 1499–1509. [Google Scholar] [CrossRef]
- Coelho, T.R.; Gaspar, R.; Figueiredo, P.; Mendonça, C.; Lazo, P.A.; Almeida, L. Human JC polyomavirus in normal colorectal mucosa, hyperplastic polyps, sporadic adenomas, and adenocarcinomas in Portugal. J. Med. Virol. 2013, 85, 2119–2127. [Google Scholar] [CrossRef]
- Hori, R.; Murai, Y.; Tsuneyama, K.; Abdel-Aziz, H.O.; Nomoto, K.; Takahashi, H.; Cheng, C.M.; Kuchina, T.; Harman, B.V.; Takano, Y. Detection of JC virus DNA sequences in colorectal cancers in Japan. Virchows Arch. 2005, 447, 723–730. [Google Scholar] [CrossRef]
- Laghi, L.; Randolph, A.E.; Chauhan, D.P.; Marra, G.; Major, E.O.; Neel, J.V.; Boland, C.R. JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 7484–7489. [Google Scholar] [CrossRef] [Green Version]
- Jung, W.T.; Li, M.S.; Goel, A.; Boland, C.R. JC virus T-antigen expression in sporadic adenomatous polyps of the colon. Cancer 2008, 112, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Hashida, Y.; Nakajima, K.; Nakajima, H.; Shiga, T.; Tanaka, M.; Murakami, M.; Matsuzaki, S.; Naganuma, S.; Kuroda, N.; Seki, Y.; et al. High load of Merkel cell polyomavirus DNA detected in the normal skin of Japanese patients with Merkel cell carcinoma. J. Clin. Virol. 2016, 82, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Vajdic, C.M.; Law, M.; Amin, J.; van Leeuwen, M.; McGregor, S.; Poynten, I.M.; Templeton, D.J.; Grulich, A.E. Incidence and time trends of anal cancer among people living with HIV in Australia. AIDS 2019, 33, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Colón-López, V.; Shiels, M.S.; Machin, M.; Ortiz, A.P.; Strickler, H.; Castle, P.E.; Pfeiffer, R.M.; Engels, E.A. Anal Cancer Risk Among People with HIV Infection in the United States. J. Clin. Oncol. 2018, 36, 68–75. [Google Scholar] [CrossRef]
- Grew, D.; Bitterman, D.; Leichman, C.G.; Leichman, L.; Sanfilippo, N.; Moore, H.G.; Du, K. HIV Infection Is Associated With Poor Outcomes for Patients With Anal Cancer in the Highly Active Antiretroviral Therapy Era. Dis. Colon Rectum 2015, 58, 1130–1136. [Google Scholar] [CrossRef]
- Grulich, A.E.; van Leeuwen, M.T.; Falster, M.O.; Vajdic, C.M. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet 2007, 370, 59–67. [Google Scholar] [CrossRef]
- Hernández-Ramírez, R.U.; Shiels, M.S.; Dubrow, R.; Engels, E.A. Cancer risk in HIV-infected people in the USA from 1996 to 2012: A population-based, registry-linkage study. Lancet HIV 2017, 4, e495–e504. [Google Scholar] [CrossRef]
- Panfil, A.R.; Martinez, M.P.; Ratner, L.; Green, P.L. Human T-cell leukemia virus-associated malignancy. Curr. Opin. Virol. 2016, 20, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Giam, C.Z.; Semmes, O.J. HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma—A Tale of Two Proteins: Tax and HBZ. Viruses 2016, 8, 161. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, Y.; Li, B.; Huang, J.; Wu, L.; Xu, D.; Yang, J.; He, J. Hepatitis viruses infection and risk of intrahepatic cholangiocarcinoma: Evidence from a meta-analysis. BMC Cancer 2012, 12, 289. [Google Scholar] [CrossRef] [Green Version]
- Su, F.H.; Le, T.N.; Muo, C.H.; Te, S.A.; Sung, F.C.; Yeh, C.C. Chronic Hepatitis B Virus Infection Associated with Increased Colorectal Cancer Risk in Taiwanese Population. Viruses 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Iloeje, U.H.; Yang, H.I.; Jen, C.L.; Su, J.; Wang, L.Y.; You, S.L.; Lu, S.N.; Chen, C.J. Risk of pancreatic cancer in chronic hepatitis B virus infection: Data from the REVEAL-HBV cohort study. Liver Int. 2010, 30, 423–429. [Google Scholar] [CrossRef]
- Hassan, M.M.; Li, D.; El-Deeb, A.S.; Wolff, R.A.; Bondy, M.L.; Davila, M.; Abbruzzese, J.L. Association between hepatitis B virus and pancreatic cancer. J. Clin. Oncol. 2008, 26, 4557–4562. [Google Scholar] [CrossRef]
- Tokita, H.; Murai, S.; Kamitsukasa, H.; Yagura, M.; Harada, H.; Takahashi, M.; Okamoto, H. High TT virus load as an independent factor associated with the occurrence of hepatocellular carcinoma among patients with hepatitis C virus-related chronic liver disease. J. Med. Virol. 2002, 67, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, V.; Malecki, M.; Tillmann, R.L.; Brockmann, M.; Schildgen, O. The Human Bocavirus Is Associated with Some Lung and Colorectal Cancers and Persists in Solid Tumors. PLoS ONE 2013, 8, e68020. [Google Scholar]
- Höpken, M.; Förster, I.; Maune, S.; Brockmann, M.; Schildgen, O.; Schildgen, V. Association of the Human Bocavirus With Tonsil Squamous Cell Carcinomas. Front. Microbiol. 2018, 9, 2450. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.; et al. Alterations in Enteric Virome Are Associated with Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [Green Version]
- Buitrago-Pérez, A.; Garaulet, G.; Vázquez-Carballo, A.; Paramio, J.M.; García-Escudero, R. Molecular Signature of HPV-Induced Carcinogenesis: pRb, p53 and Gene Expression Profiling. Curr. Genomics. 2009, 10, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, M.; D’Abramo, C.M.; Archambault, J. Molecular Mechanisms of Human Papillomavirus-Induced Carcinogenesis. Public Health Genom. 2009, 12, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Emlet, C.; Ruffin, M.; Lamendella, R. Enteric Virome and Carcinogenesis in the Gut. Dig. Dis. Sci. 2020, 65, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Braaten, K.P.; Laufer, M.R. Human Papillomavirus (HPV), HPV-Related Disease, and the HPV Vaccine. Rev. Obstet. Gynecol. 2008, 1, 2–10. [Google Scholar]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.J. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Filippakis, H.; Spandidos, D.A.; Sourvinos, G. Herpesviruses: Hijacking the Ras signaling pathway. Biochim. Biophys. Acta 2010, 1803, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Xiao, B.; Si, H.; Cervini, A.; Gao, J.; Lu, J.; Upadhyay, S.K.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012, 8, e1002566. [Google Scholar] [CrossRef] [Green Version]
- Frazer, I.H. The actinic keratosis virome: Can we prevent squamous cell carcinoma with a vaccine? Curr. Probl. Dermatol. 2015, 46, 28–35. [Google Scholar]
- Cassler, N.M.; Merrill, D.; Bichakjian, C.K.; Brownell, I. Merkel Cell Carcinoma Therapeutic Update. Curr. Treat. Options Oncol. 2016, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research (IWMCC) Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Khalili, K. Polyomaviruses and human cancer: Molecular mechanisms underlying patterns of tumorigenesis. Virology 2004, 324, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef]
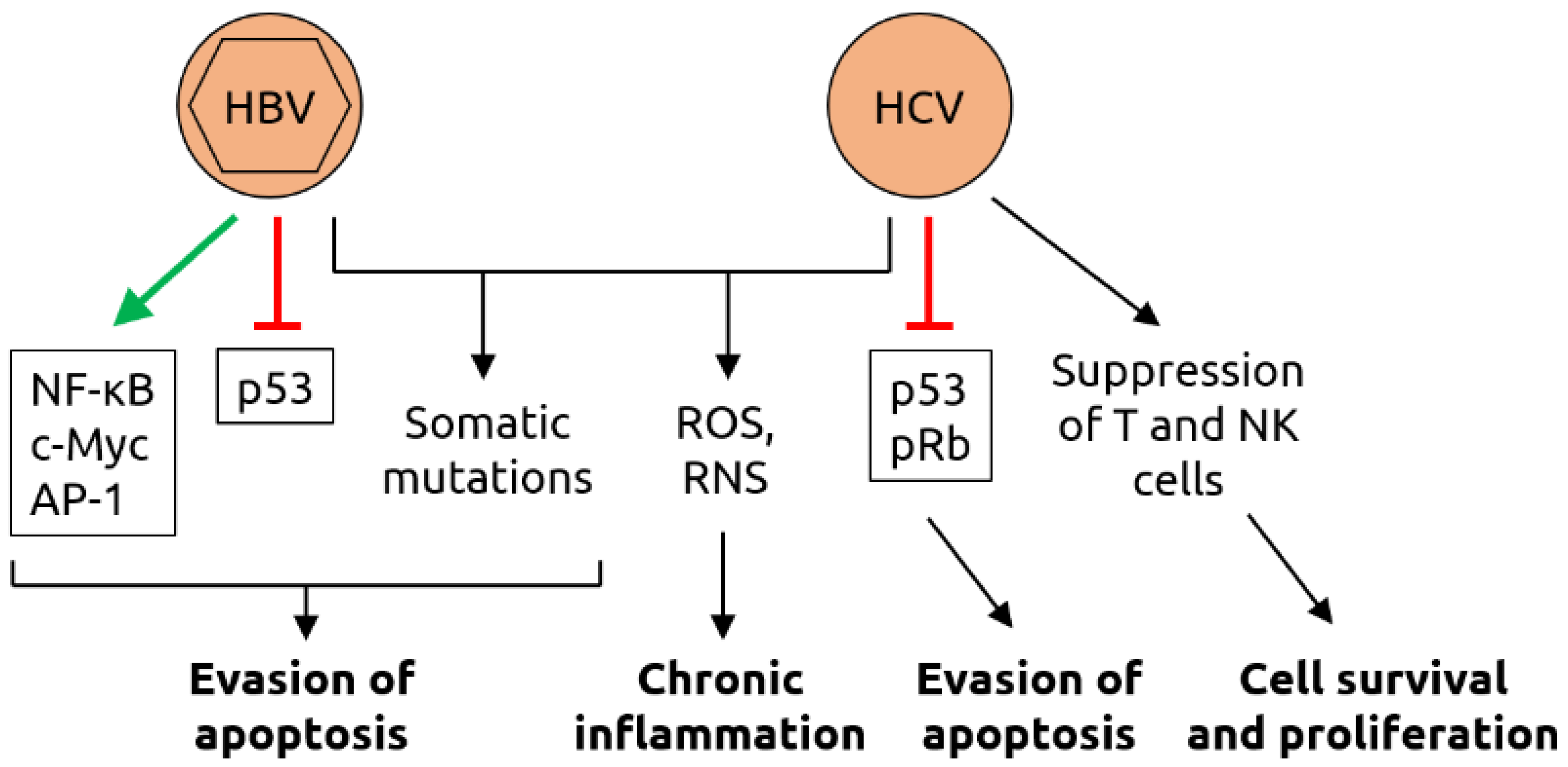
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpiński, T.M. The Microbiota and Pancreatic Cancer. Gastroenterol. Clin. North Am. 2019, 48, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Matijašić, M.; Meštrović, T.; Paljetak, H.Č.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T., 4th; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, e02248-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handley, S.A.; Devkota, S. Going Viral: A Novel Role for Bacteriophage in Colorectal Cancer. mBio 2019, 10, e02626-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, K.J.; Baxter, N.T.; Schloss, P.D. Metabolic and Community Synergy of Oral Bacteria in Colorectal Cancer. mSphere 2016, 1, e00102-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Stripling, J.; Rodriguez, M. Current Evidence in Delivery and Therapeutic Uses of Fecal Microbiota Transplantation in Human Diseases-Clostridium difficile Disease and Beyond. Am. J. Med. Sci. 2018, 356, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Kube, M.; Klumpp, J.; Schuppler, M.; Biedermann, L.; Hecht, J.; Hombach, M.; Keller, P.M.; Rogler, G.; Moelling, K. Analysis of the intestinal microbiome of a recovered Clostridium difficile patient after fecal transplantation. Digestion 2013, 88, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, D.H.; Roach, B.; Walter, J.; Lobenberg, R.; Wong, K. A51 Effect of lyophilized sterile fecal filtrate vs lyophilized donor stool on recurrent Clostridium difficile infection (RCDI): Preliminary results from a randomized, double-blind pilot study. J. Can. Assoc. Gastroenterol. 2019, 2 (Suppl. 2), 101–102. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.L.; Chan, P.K.S.; Chan, M.C.W.; Wu, J.C.Y.; et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018, 67, 634–643. [Google Scholar]
- Broecker, F.; Russo, G.; Klumpp, J.; Moelling, K. Stable core virome despite variable microbiome after fecal transfer. Gut Microbes 2017, 8, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Draper, L.A.; Ryan, F.J.; Smith, M.K.; Jalanka, J.; Mattila, E.; Arkkila, P.A.; Ross, R.P.; Satokari, R.; Hill, C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018, 6, 220. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Aarnoutse, R.; Ziemons, J.; Penders, J.; Rensen, S.S.; de Vos-Geelen, J.; Smidt, M.L. The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4145. [Google Scholar] [CrossRef] [Green Version]
- Fluckiger, A.; Daillère, R.; Sassi, M.; Sixt, B.S.; Liu, P.; Loos, F.; Richard, C.; Rabu, C.; Alou, M.T.; Goubet, A.G.; et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020, 369, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur. J. Immunol. 2015, 45, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H.; Bund, T.; de Villiers, E.M. Infectious Agents in Bovine Red Meat and Milk and Their Potential Role in Cancer and Other Chronic Diseases. Curr. Top. Microbiol. Immunol. 2017, 407, 83–116. [Google Scholar]
- Arroyo Mühr, L.S.; Bzhalava, Z.; Hortlund, M.; Lagheden, C.; Nordqvist Kleppe, S.; Bzhalava, D.; Hultin, E.; Dillner, J. Viruses in cancers among the immunosuppressed. Int. J. Cancer 2017, 141, 2498–2504. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, C.R.; Amano, H.; Ueda, Y.; Qin, J.; Miyamura, T.; Suzuki, T.; Li, X.; Barrett, J.W.; McFadden, G. Complete genomic sequence and comparative analysis of the tumorigenic poxvirus Yaba monkey tumor virus. J. Virol. 2003, 77, 13335–13347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehoud, C.; Dryga, A.; Hwang, Y.; Nagy-Szakal, D.; Hollister, E.B.; Luna, R.A.; Versalovic, J.; Kellermayer, R.; Bushman, F.D. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. mBio 2016, 7, e00322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunse, A.; Deng, L.; Pan, X.; Hui, Y.; Castro-Mejía, J.L.; Kot, W.; Nguyen, D.N.; Secher, J.B.; Nielsen, D.S.; Thymann, T. Fecal filtrate transplantation protects against necrotizing enterocolitis. ISME J. 2021. Epub ahead of print. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family | Virus | Cancer Type | Observations |
|---|---|---|---|
| Papillomaviridae | HPV-16 | Anal | HPV, especially HPV-16, is a possible risk factor for anal and rectal cancer [8,9] and a significant prognostic marker, especially for locally advanced disease [10] |
| HPV | Bladder | HPV (different serotypes) may be linked to bladder cancer in a small number of cases [11] | |
| HPV-16, -18 | Cervical | Association between infection with high-risk HPV serotype (mainly HPV-16 and-18) and development of cervical cancer [11,12,13] | |
| HPV-18 | Colorectal | HPV, especially HPV-18, is a possible risk factor for colorectal cancer [14,15], however, another study found no association [16] | |
| HPV-16, -18, -26, -57 | Esophageal | HPV-16 is a risk factor for esophageal carcinoma [17,18]; HPV infection (mainly HPV-16, -18, -26 and-57) is common in esophageal carcinoma [19] | |
| HPV-16 | Head and neck (SCC) | HPV infection, especially HPV-16, is associated with head and neck cancer [11,20,21] and better long-term outcome [22] | |
| HPV-6 | Oral | Association of HPV-6 with oral cancer [23] | |
| HPV-16 | Prostate | Association of HPV-16 with prostate cancer [24] | |
| HPV-16, -18, -58 | Renal | Association of HPV-16, -18 and-58 with renal cell carcinoma [25] | |
| HPV-5, -8 | Skin and mucosal | Papillomavirus DNA frequently detected in skin-and mucosa-associated cancers [26]; HPV-5 and-8 are associated with epidermodysplasia verruciformis associated with a high risk of skin cancer [27,28] | |
| HPV-16 | Vulvar | Association between HPV, especially HPV-16, and vulvar squamous cell carcinoma [29] | |
| Herpesviridae | CMV (HHV5) | Colorectal | CMV DNA is more abundant cancer tissues compared to healthy tissues [30]; CMV-positive tumors in non-elderly patients are associated with increased disease-free survival rate [31]; specific genetic polymorphisms of CMV are linked to different clinical outcomes [32] |
| EBV (HHV4) | Colorectal | Possible association of EBV with colorectal carcinoma [33], however, no association found in another study [16] | |
| EBV (HHV4) | Esophageal | EBV is associated with esophageal cancer [34] | |
| EBV (HHV4) | Gastric | Possible involvement of EBV in gastric cancer and precursor lesions [35]; patients with EBV-positive gastric cancer had a better response to chemotherapy and better survival [36] | |
| EBV (HHV4) | Hepatic | EBV infections detected in HCC tissues [37] | |
| EBV (HHV4) | Lymphoma (Burkitt) | EBV infections contribute to Burkitt lymphoma [38] | |
| EBV (HHV4) | Lymphoma (DLBCL) | EBV RNA detected in B-cell lymphoma samples [39] | |
| EBV (HHV4) | Lymphoma (PTCL) | EBV expression associated with some subtypes of peripheral T-cell lymphomas [40] | |
| EBV (HHV4) | Oral | Higher proportion of EBV-positive oral squamous cell carcinoma in industrialized countries [41] | |
| EBV (HHV4) | Skin and mucosal | EBV DNA frequently detected in skin and mucosal cancers [26] | |
| HHV6 | Lymphoma (DLBCL) | HHV6 RNA detected in B-cell lymphoma samples [39] | |
| HHV6 | Malignant melanoma | HHV6 DNA frequently detected in malignant melanoma [26] | |
| HHV7 | Bladder | HHV7 DNA frequently detected in bladder cancer [26] | |
| HHV7 | Lymphoma (CTCL) | HHV7 DNA frequently detected in cutaneous T-cell lymphoma (Mycosis fungoides) [26] | |
| HHV7 | Oral | HHV7 DNA frequently detected in oral cavity cancer [26] | |
| HSV (HHV1/2) | Oral | Higher proportion of HSV-positive oral squamous cell carcinoma in industrialized countries [41] | |
| KSHV (HHV8) | Kaposi sarcoma | In HIV-infected individuals, KSHV infection is associated with Kaposi sarcoma [42] | |
| Polyomaviridae | BKV | Bladder | Possible association of BKV with bladder cancer [26] |
| BKV | Colorectal | Possible association of BKV with colorectal cancer [43,44], however, other studies found no association [16,45] | |
| JCV | Colorectal | JCV is associated with colorectal cancer [45,46] and may be involved in carcinogenesis [47], specifically in chromosomal instability [48]; JCV T-antigen is expressed in early-stage colorectal cancer [49], however, another study found no association [16] | |
| MCV | Merkel cell carcinoma | MCV is the major causative factor for Merkel cell carcinoma [50,51] | |
| Retroviridae | HIV | Anal | HIV-positive people have increased risk for anal cancer [52,53] and worse overall colostomy-free survival rates [54] |
| HIV | Cervical | Cervical cancer is more prevalent in HIV-positive individuals, likely because of increased susceptibility to HPV infection [55,56] | |
| HIV | Kaposi sarcoma | Kaposi sarcoma is more prevalent in HIV-positive individuals, likely because of increased susceptibility to KSHV infection [55,56] | |
| HIV | Lymphoma (NHL) | Aggressive B cell non-Hodgkin lymphoma is more prevalent in HIV-positive individuals, likely because of increased susceptibility to EBV infection [55,56] | |
| HTLV-1 | Lymphoma (ATLL) | HTLV-1 induces adult T-cell leukemia/lymphoma in 5% of infected individuals [57] through random integration into the host genome [58] | |
| Others | HBV | Bile duct | HBV is a risk factor for bile duct cancer [59] |
| HBV | Colorectal | Chronic HBV infection is a risk factor for colorectal cancer [60] | |
| HBV | Hepatic | Liver cancer is associated with HBV [11] | |
| HBV | Pancreatic | Chronic HBV infection [61] or past exposure [62] are risk factors for pancreatic cancer | |
| HCV | Bile duct | HCV is a risk factor for bile duct cancer [59] | |
| HCV | Hepatic | Liver cancer is associated with HCV [11] | |
| TTV | Hepatic | TTV is a risk factor for hepatocellular carcinoma [63] | |
| HBoV | Colorectal | Some colorectal cancers are associated with HBoV [64] | |
| HBoV | Lung | Some lung cancers are associated with HBoV [64] | |
| HBoV | Tonsillar | Association of HBoV with tonsil squamous cell carcinoma [65] | |
| Orthobunyaviruses | Colorectal | High abundance of orthobunyaviruses in colorectal cancer [66] | |
| Parvoviruses | Skin | Parvovirus DNA frequently detected in skin-associated cancers [26] | |
| Anelloviruses | Mucosal | Anellovirus DNA frequently detected in mucosal cancers [26] | |
| Anelloviruses | Leukemias | Anellovirus DNA frequently detected in leukemias [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broecker, F.; Moelling, K. The Roles of the Virome in Cancer. Microorganisms 2021, 9, 2538. https://doi.org/10.3390/microorganisms9122538
Broecker F, Moelling K. The Roles of the Virome in Cancer. Microorganisms. 2021; 9(12):2538. https://doi.org/10.3390/microorganisms9122538
Chicago/Turabian StyleBroecker, Felix, and Karin Moelling. 2021. "The Roles of the Virome in Cancer" Microorganisms 9, no. 12: 2538. https://doi.org/10.3390/microorganisms9122538
APA StyleBroecker, F., & Moelling, K. (2021). The Roles of the Virome in Cancer. Microorganisms, 9(12), 2538. https://doi.org/10.3390/microorganisms9122538