
The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges

{kind=link}
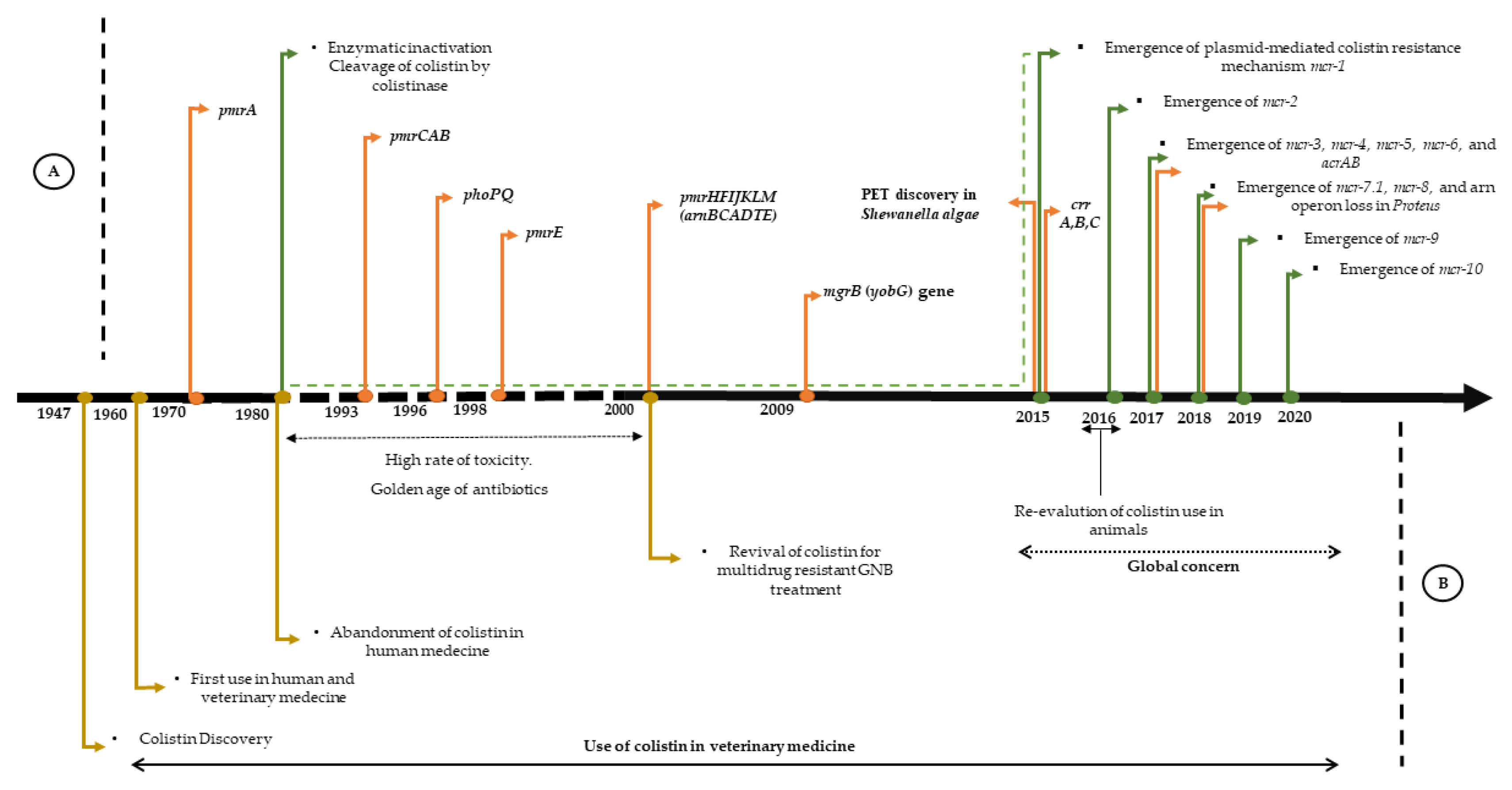{kind=link}
Abstract
:1. Introduction
2. Pathways Leading to Colistin Action and Mechanisms of Resistance
2.1. Mode of Action
2.2. Mechanisms of Resistance
3. From Electron Microscopy to the Discovery of Regulatory Genes
3.1. Intrinsic Resistance Bacteria
3.2. Enzymatic Inactivation
3.3. Regulatory System Discovery
3.4. Arn Operon Discovery
3.5. mgrB (yobG) Discovery
3.6. CrrAB Discovery
4. Exploitation of Colistin Resistance: Methods and Challenges
4.1. Lipid A Extraction and Mass Spectrometry
4.2. The Discovery of New Variants and Mutations in a Common Gene
4.3. Genetic Toolbox and Colistin Resistance Research
4.4. Whole Genome Sequencing: A Modern Approach for a Growing Challenge
4.5. Transcriptomics and Proteomics
4.6. Application of CRISPR-cas 9-Based Genome Editing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baron, S.; Hadjadj, L.; Rolain, J.M.; Olaitan, A.O. Molecular mechanisms of polymyxin resistance: Knowns and unknowns. Int. J. Antimicrob. Agents 2016, 48, 583–591. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Texte, I.P.A. du Annales de l’Institut Pasteur: Journal de Microbiologie/Publiées sous le Patronage de M. Pasteur par E. Duclaux. 1961. Available online: https://gallica.bnf.fr/ark:/12148/cb34348753q/date (accessed on 19 February 2021).
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Stansly, P.G.; Schlosser, M.E. Studies on Polymyxin: Isolation and Identification of Bacillus polymyxa and Differentiation of Polymyxin from Certain Known Antibiotics. J. Bacteriol. 1947, 54, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, E.S.M.A.E.G.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, K.S.; Pogue, J.M.; Tran, T.B.; Nation, R.L.; Li, J. Agents of Last Resort: Polymyxin Resistance. Infect. Dis. Clin. North Am. 2016, 30, 391–414. [Google Scholar] [CrossRef] [PubMed]
- Sekyere, O.J.; Govinden, U.; Bester, L.A.; Essack, S.Y. Colistin and tigecycline resistance in carbapenemase-producing Gram-negative bacteria: Emerging resistance mechanisms and detection methods. J. Appl. Microbiol. 2016, 121, 601–617. [Google Scholar] [CrossRef]
- Haenni, M.; Poirel, L.; Kieffer, N.; Châtre, P.; Saras, E.; Métayer, V.; Dumoulin, R.; Nordmann, P.; Madec, J.Y. Co-occurrence of extended spectrum lactamase and MCR-1 encoding genes on plasmids. Lancet Infect. Dis. 2016, 16, 281–282. [Google Scholar] [CrossRef] [Green Version]
- Lopes, J.; Inniss, W.E. Electron microscopy of effect of polymyxin on Escherichia coli lipopolysaccharide. J. Bacteriol. 1969, 100, 1128–1129. [Google Scholar] [CrossRef] [Green Version]
- Koike, M.; Iida, K.; Matsuo, T. Electron microscopic studies on mode of action of polymyxin. J. Bacteriol. 1969, 97, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Storm, D.R.; Rosenthal, K.S.; Swanson, P.E. Polymyxin and related peptide antibiotics. Annu. Rev. Biochem. 1977, 46, 723–763. [Google Scholar] [CrossRef] [PubMed]
- Newton, B.A. Site of action of polymyxin on Pseudomonas aeruginosa: Antagonism by cations. J. Gen. Microbiol. 1954, 10, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, P.R.G.; Teuber, M. Action of polymyxin B on bacterial membranes: Morphological changes in the cytoplasm and in the outer membrane of Salmonella typhimurium and Escherichia coli B. Antimicrob. Agents Chemother. 1975, 8, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, B.A. The properties and mode of action of the polymyxins. Microbiol. Mol. Biol. Rev. 1956, 20, 14. [Google Scholar] [CrossRef] [Green Version]
- Few, A.V. The interaction of polymyxin E with bacterial and other lipids. BBA-Biochim. Biophys. Acta 1955, 16, 137–145. [Google Scholar] [CrossRef]
- Petersdorf, R.G.; Plorde, J.J. Colistin-A Reappraisal. JAMA J. Am. Med. Assoc. 1963, 183, 123–125. [Google Scholar] [CrossRef]
- Conrad, R.S.; Wulf, R.G.; Clay, D.L. Effects of carbon sources of antibiotic resistance in pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1979, 15, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Haas, G.J.; Sevag, M.G. Critical role of amino acids on the sensitivity and development of resistance to polymyxin B. Arch. Biochem. Biophys. 1953, 43, 11–24. [Google Scholar] [CrossRef]
- Shimizu, S.; Iyobe, S.; Mitsuhashi, S. Inducible high resistance to colistin in Proteus strains. Antimicrob. Agents Chemother. 1977, 12, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Sud, I.J.; Feingold, D.S. Mechanism of polymyxin B resistance in Proteus mirabilis. J. Bacteriol. 1970, 104, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.; Nadakavukaren, M.; Tsang, J. Electron microscopic observations of polysaccharide components in polymyxin b treated outer membranes from serratia marcescens. J. Antibiot. (Tokyo) 1979, 32, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Mise en evidence des polysaccharides sur coupes fines en microscopie electronique. J. Microsc. 1967, 6, 987–1018. [Google Scholar]
- Weber, D.A.; Nadakavukaren, M.J.; Tsang, J.C. Localization of polysaccharide components in polymyxin b treated cells of serra tia marcescens. J. Antibiot. (Tokyo) 1978, 31, 732–735. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.D.; Iannetta, A.; Wedgwood, R.J. Activity of colistin against pseudomonas aeruginosa: Inhibition by calcium. J. Infect. Dis. 1971, 124, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W. Alterations in structure of the cell envelope. Ann. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.A.; Rolain, J.M. Efflux pump inhibitor CCCP to rescue colistin susceptibility in mcr-1 plasmid-mediated colistin-resistant strains and Gram-negative bacteria. J. Antimicrob. Chemother. 2018, 73, 1862–1871. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, I.M.; Koyama, Y. Selective cleavage of a peptide antibiotic, colistin by colistinase. J. Antibiot. (Tokyo) 1980, 33, 1551–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffatt, J.H.; Harper, M.; Boyce, J.D. Mechanisms of Polymyxin Resistance. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1145, pp. 55–71. [Google Scholar] [CrossRef]
- Yin, J.; Wang, G.; Cheng, D.; Fu, J.; Qiu, J.; Yu, Z. Inactivation of polymyxin by hydrolytic mechanism. Antimicrob. Agents Chemother. 2019, 63, 2378–2396. [Google Scholar] [CrossRef] [Green Version]
- Czub, M.P.; Zhang, B.; Chiarelli, M.P.; Majorek, K.A.; Joe, L.; Porebski, P.J.; Revilla, A.; Wu, W.; Becker, D.P.; Minor, W.; et al. A Gcn5-Related N-Acetyltransferase (GNAT) Capable of Acetylating Polymyxin B and Colistin Antibiotics in Vitro. Biochemistry 2018, 57, 7011–7020. [Google Scholar] [CrossRef]
- Burckhardt, R.M.; Semerena, E.J.C. Small-Molecule Acetylation by GCN5-Related N -Acetyltransferases in Bacteria. Microbiol. Mol. Biol. Rev. 2020, 84. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, H.P.; Sarvas, M.; Calcagno, S.; Lounatmaa, K. Isolation and genetic characterization of polymyxin-resistant mutants of Salmonella. FEMS Microbiol. Lett. 1978, 3, 323–326. [Google Scholar] [CrossRef]
- Vaara, M.; Vaara, T.; Sarvas, M. Decreased binding of polymyxin by polymyxin-resistant mutants of Salmonella typhimurium. J. Bacteriol. 1979, 139, 664–667. [Google Scholar] [CrossRef] [Green Version]
- Gunn, J.S.; Miller, S.I. PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J. Bacteriol. 1996, 178, 6857–6864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaara, M. Increased outer membrane resistance to ethylenediaminetetraacetate and cations in novel lipid A mutants. J. Bacteriol. 1981, 148, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Roland, K.L.; Martin, L.E.; Esther, C.R.; Spitznagel, J.K. Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two- component regulatory system with a possible role in virulence. J. Bacteriol. 1993, 175, 4154–4164. [Google Scholar] [CrossRef] [Green Version]
- Gunn, J.S. The Salmonella PmrAB regulon: Lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 2008, 16, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hsu, F.F.; Turk, J.; Groisman, E.A. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J. Bacteriol. 2004, 186, 4124–4133. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.I.; Kukral, A.M.; Mekalanos, J.J. A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc. Natl. Acad. Sci. USA 1989, 86, 5054–5058. [Google Scholar] [CrossRef] [Green Version]
- Kier, L.D.; Weppelman, R.M.; Ames, B.N. Regulation of nonspecific acid phosphatase in Salmonella: phoN and phoP genes. J. Bacteriol. 1979, 138, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groisman, E.A. The pleiotropic two-component regulatory system PhoP-PhoQ. J. Bacteriol. 2001, 183, 1835–1842. [Google Scholar] [CrossRef] [Green Version]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E.W. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Gunn, J.S.; Bainbridge, B.; Darveau, R.P.; Hackett, M.; Miller, S.I. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science 1997, 276, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S.; Lim, K.B.; Krueger, J.; Kim, K.; Guo, L.; Hackett, M.; Miller, S.I. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 1998, 27, 1171–1182. [Google Scholar] [CrossRef]
- Gunn, J.S.; Ryan, S.S.; Van Velkinburgh, J.C.; Ernst, R.K.; Miller, S.I. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 2000, 68, 6139–6146. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.; Leulmi, Z.; Villard, C.; Olaitan, A.O.; Telke, A.A.; Rolain, J.M. Inactivation of the arn operon and loss of aminoarabinose on lipopolysaccharide as the cause of susceptibility to colistin in an atypical clinical isolate of proteus vulgaris. Int. J. Antimicrob. Agents 2018, 51, 450–457. [Google Scholar] [CrossRef]
- Kato, A.; Tanabe, H.; Utsumi, R. Molecular characterization of the PhoP-PhoQ two-component system in Escherichia coli K-12: Identification of extracellular Mg2+-responsive promoters. J. Bacteriol. 1999, 181, 5516–5520. [Google Scholar] [CrossRef] [Green Version]
- Lippa, A.M.; Goulian, M. Feedback inhibition in the PhoQ/PhoP signaling system by a membrane peptide. PLoS Genet. 2009, 5, e1000788. [Google Scholar] [CrossRef] [Green Version]
- Hemm, M.R.; Paul, B.J.; Schneider, T.D.; Storz, G.; Rudd, K.E. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 2008, 70, 1487–1501. [Google Scholar] [CrossRef] [Green Version]
- Mouna, H.; Stylianos, C.; Linda, H.; Efthimia, P.; Sophia, P.; Nikoletta, C.; Sophia, T.; Vassiliki, P.; Nikoletta, S.; Iris, S.; et al. Inactivation of mgrB gene regulator and resistance to colistin is becoming endemic in carbapenem-resistant Klebsiella pneumoniae in Greece: A nationwide study from 2014 to 2017. Int. J. Antimicrob. Agents 2020, 55, 5930. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Diene, S.M.; Kempf, M.; Berrazeg, M.; Bakour, S.; Gupta, S.K.; Thongmalayvong, B.; Akkhavong, K.; Somphavong, S.; Paboriboune, P.; et al. Worldwide emergence of colistin resistance in Klebsiella pneumoniae from healthy humans and patients in Lao PDR, Thailand, Israel, Nigeria and France owing to inactivation of the PhoP/PhoQ regulator mgrB: An epidemiological and molecular study. Int. J. Antimicrob. Agents 2014, 44, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, M.S.; Suzuki, Y.; Jones, M.B.; Marshall, S.H.; Rudin, S.D.; Van Duin, D.; Kaye, K.; Jacobs, M.R.; Bonomo, R.A.; Adamsa, M.D. Genomic and transcriptomic analyses of colistin-resistant clinical isolates of Klebsiella pneumoniae reveal multiple pathways of resistance. Antimicrob. Agents Chemother. 2015, 59, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Bardet, L.; Rolain, J.M. Development of new tools to detect colistin-resistance among enterobacteriaceae strains. Can. J. Infect. Dis. Med. Microbiol. 2018, 2018. [Google Scholar] [CrossRef]
- Welker, M.; Van Belkum, A.; Girard, V.; Charrier, J.P.; Pincus, D. An update on the routine application of MALDI-TOF MS in clinical microbiology. Expert Rev. Proteom. 2019, 16, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.; Popkova, Y.; Engel, K.; Schiller, J. Recent Developments of Useful MALDI Matrices for the Mass Spectrometric Characterization of Lipids. Biomolecules 2018, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Amano, J.; Sugahara, D.; Osumi, K.; Tanaka, K.K. Negative-ion MALDI-QIT-TOFMSn for structural determination of fucosylated and sialylated oligosaccharides labeled with a pyrene derivative. Glycobiology 2009, 19, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maumus, L.G.; Clements, A.; Filloux, A.; McCarthy, R.R.; Mostowy, S. Direct detection of lipid A on intact Gram-negative bacteria by MALDI-TOF mass spectrometry. J. Microbiol. Methods 2016, 120, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Dortet, L.; Potron, A.; Bonnin, R.A.; Plesiat, P.; Naas, T.; Filloux, A.; Maumus, L.G. Rapid detection of colistin resistance in Acinetobacter baumannii using MALDI-TOF-based lipidomics on intact bacteria. Sci. Rep. 2018, 8, s41598–s41618. [Google Scholar] [CrossRef]
- Furniss, R.C.D.; Dortet, L.; Bolland, W.; Drews, O.; Sparbier, K.; Bonnin, R.A.; Filloux, A.; Kostrzewa, M.; Mavridou, D.A.I.; Maumus, L.G. Detection of colistin resistance in Escherichia coli by use of the MALDI biotyper sirius mass spectrometry system. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [Green Version]
- Dortet, L.; Broda, A.; Bernabeu, S.; Glupczynski, Y.; Bogaerts, P.; Bonnin, R.; Naas, T.; Filloux, A.; Maumus, L.G. Optimization of the MALDIxin test for the rapid identification of colistin resistance in Klebsiella pneumoniae using MALDI-TOF MS. J. Antimicrob. Chemother. 2020, 75, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telke, A.A.; Rolain, J.M. Functional genomics to discover antibiotic resistance genes: The paradigm of resistance to colistin mediated by ethanolamine phosphotransferase in Shewanella algae MARS 14. Int. J. Antimicrob. Agents 2015, 46, 648–652. [Google Scholar] [CrossRef]
- Hadjadj, L.; Baron, S.A.; Diene, S.M.; Rolain, J.M. How to discover new antibiotic resistance genes? Expert Rev. Mol. Diagn. 2019, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- McClure, E.E.; Chávez, A.S.O.; Shaw, D.K.; Carlyon, J.A.; Ganta, R.R.; Noh, S.M.; Wood, D.O.; Bavoil, P.M.; Brayton, K.A.; Martinez, J.J.; et al. Engineering of obligate intracellular bacteria: Progress, challenges and paradigms. Nat. Rev. Microbiol. 2017, 15, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Kulasekara, H.D. Transposon mutagenesis. Methods Mol. Biol. 2014, 1149, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Way, J.C.; Davis, M.A.; Morisato, D.; Roberts, D.E.; Kleckner, N. New Tn10 derivatives for transposon mutagenesis and for construction of lacZ operon fusions by transposition. Gene 1984, 32, 369–379. [Google Scholar] [CrossRef]
- Ruvkun, G.B.; Ausubel, F.M. A general method for site-directed mutagenesis in prokaryotes. Nature 1981, 289, 85–88. [Google Scholar] [CrossRef]
- Hayes, F. Transposon-Based Strategies for Microbial Functional Genomics and Proteomics. Annu. Rev. Genet. 2003, 37, 3–29. [Google Scholar] [CrossRef]
- Telke, A.A.; Olaitan, A.O.; Morand, S.; Rolain, J.M. SoxRS induces colistin hetero-resistance in Enterobacter asburiae and Enterobacter cloacae by regulating the acrAB-tolC efflux pump. J. Antimicrob. Chemother. 2017, 72, 2715–2721. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Feng, Y.; Zong, Z. Heterogeneous resistance to colistin in Enterobacter cloacae complex due to a new small transmembrane protein. J. Antimicrob. Chemother. 2019, 74, 2551–2558. [Google Scholar] [CrossRef] [PubMed]
- Lampe, D.J.; Akerley, B.J.; Rubin, E.J.; Mekalanos, J.J.; Robertson, H.M. Hyperactive transposase mutants of the Himar1 mariner transposon. Proc. Natl. Acad. Sci. USA 1999, 96, 11428–11433. [Google Scholar] [CrossRef] [Green Version]
- Rubin, E.J.; Akerley, B.J.; Novik, V.N.; Lampe, D.J.; Husson, R.N.; Mekalanos, J.J. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. USA 1999, 96, 1645–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schürch, A.C.; van Schaik, W. Challenges and opportunities for whole-genome sequencing-based surveillance of antibiotic resistance. Ann. N. Y. Acad. Sci. 2017, 1388, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.; Walsh, F. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol. Ecol. 2016, 92, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y. Transferability of MCR-1/2 Polymyxin Resistance: Complex Dissemination and Genetic Mechanism. ACS Infect. Dis. 2018, 4, 291–300. [Google Scholar] [CrossRef]
- Khedher, M.B.; Baron, S.A.; Riziki, T.; Ruimy, R.; Raoult, D.; Diene, S.M.; Rolain, J.M. Massive analysis of 64,628 bacterial genomes to decipher water reservoir and origin of mobile colistin resistance genes: Is there another role for these enzymes? Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, C.; Zhang, R.; Chen, Y.; Shen, Y.; Hu, F.; Liu, D.; Lu, J.; Guo, Y.; Xia, X.; et al. Changes in colistin resistance and mcr-1 abundance in Escherichia coli of animal and human origins following the ban of colistin-positive additives in China: An epidemiological comparative study. Lancet Infect. Dis. 2020, 20, 1161–1171. [Google Scholar] [CrossRef]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Singh, K.S.; Butaye, P.; Goossens, H.; Kumar, M.S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 280. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Li, H.; Shen, Y.; Liu, Z.; Wang, S.; Shen, Z.; Zhang, R.; Walsh, T.R.; Shen, J.; Wang, Y. Novel plasmid-mediated colistin resistance gene mcr-3 in Escherichia coli. MBio 2017, 8, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Villa, L.; Feudi, C.; Curcio, L.; Orsini, S.; Luppi, A.; Pezzotti, G.; Magistrali, C.F. Novel plasmid-mediated colistin resistance mcr-4 gene in Salmonella and Escherichia coli, Italy 2013, Spain and Belgium, 2015 to 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Borowiak, M.; Fischer, J.; Hammerl, J.A.; Hendriksen, R.S.; Szabo, I.; Malorny, B. Identification of a novel transposon-associated phosphoethanolamine transferase gene, mcr-5, conferring colistin resistance in d-tartrate fermenting Salmonella enterica subsp. enterica serovar Paratyphi B. J. Antimicrob. Chemother. 2017, 72, 3317–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Q.; Li, Y.X.; Lei, C.W.; Zhang, A.Y.; Wang, H.N. Novel plasmid-mediated colistin resistance gene mcr-7.1 in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1791–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Li, J.; Yin, W.; Wang, S.; Zhang, S.; Shen, J.; Shen, Z.; Wang, Y. Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerg. Microbes Infect. 2018, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Carroll, L.M.; Gaballa, A.; Guldimann, C.; Sullivan, G.; Henderson, L.O.; Wiedmann, M. Identification of novel mobilized colistin resistance gene mcr-9 in a multidrug-resistant, colistin-susceptible salmonella enterica serotype typhimurium isolate. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Feng, Y.; Liu, L.; Wei, L.; Kang, M.; Zong, Z. Identification of novel mobile colistin resistance gene mcr-10. Emerg. Microbes Infect. 2020, 9, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Hadjadj, L.; Baron, S.A.; Olaitan, A.O.; Morand, S.; Rolain, J.M. Co-occurrence of Variants of mcr-3 and mcr-8 Genes in a Klebsiella pneumoniae Isolate from Laos. Front. Microbiol. 2019, 10, 2720. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, A.; Ruiz, U.M.; Hernández, M.; Villoldo, M.P.; Lázaro, R.D.; Domínguez, L.; Quesada, A. Involvement of hpap2 and dgkA Genes in Colistin Resistance Mediated by mcr Determinants. Antibiotics 2020, 9, 531. [Google Scholar] [CrossRef]
- Barcz, A.K.; Gomez, J.E.; Kaufmann, B.B.; Hinson, E.R.; Cosimi, L.; Borowsky, M.L.; Onderdonk, A.B.; Stanley, S.A.; Kaur, D.; Bryant, K.F.; et al. RNA signatures allow rapid identification of pathogens and antibiotic susceptibilities. Proc. Natl. Acad. Sci. USA 2012, 109, 6217–6222. [Google Scholar] [CrossRef] [Green Version]
- Dunne, W.M.; Jaillard, M.; Rochas, O.; Van Belkum, A. Microbial genomics and antimicrobial susceptibility testing. Expert Rev. Mol. Diagn. 2017, 17, 257–269. [Google Scholar] [CrossRef]
- Guigo, R.; de Hoon, M. Recent advances in functional genome analysis. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Li, H.; Peng, X. Proteomics approach to understand bacterial antibiotic resistance strategies. Expert Rev. Proteom. 2019, 16, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Meng, Q.; Wang, Y.; Xia, G.; Xia, X.; Shen, J. Comprehensive proteomic and metabolomic profiling of mcr-1-mediated colistin resistance in Escherichia coli. Int. J. Antimicrob. Agents 2019, 53, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Vranakis, I.; Goniotakis, I.; Psaroulaki, A.; Sandalakis, V.; Tselentis, Y.; Gevaert, K.; Tsiotis, G. Proteome studies of bacterial antibiotic resistance mechanisms. J. Proteomics 2014, 97, 88–99. [Google Scholar] [CrossRef]
- Reyes, F.M.; Falcón, R.M.; Chiva, C.; Pachón, J.; Andreu, D.; Rivas, L. The cost of resistance to colistin in Acinetobacter baumannii: A proteomic perspective. Proteomics 2009, 9, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Rasmussen, P.K.; Bai, Y.; Chen, X.; Cai, T.; Wang, J.; Guo, X.; Xie, Z.; Ding, X.; Niu, L.; et al. Proteomic changes of Klebsiella pneumoniae in response to colistin treatment and crrB mutation-mediated colistin resistance. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Luo, M.L.; Leenay, R.T.; Beisel, C.L. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 2016, 113, 930–943. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 8096. [Google Scholar] [CrossRef]
- Dong, H.; Xiang, H.; Mu, D.; Wang, D.; Wang, T. Exploiting a conjugative CRISPR/Cas9 system to eliminate plasmid harbouring the mcr-1 gene from Escherichia coli. Int. J. Antimicrob. Agents 2019, 53, 1–8. [Google Scholar] [CrossRef]
- Sun, L.; He, T.; Zhang, L.; Pang, M.; Zhang, Q.; Zhou, Y.; Bao, H.; Wang, R. Generation of newly discovered resistance gene mcr-1 knockout in Escherichia coli using the CRISPR/Cas9 system. J. Microbiol. Biotechnol. 2017, 27, 1276–1280. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; McBride, S.W.; Hullahalli, K.; Palmer, K.L.; Duerkop, B.A. Conjugative delivery of CRISPR-Cas9 for the selective depletion of antibiotic-resistant enterococci. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Vercoe, R.B.; Chang, J.T.; Dy, R.L.; Taylor, C.; Gristwood, T.; Clulow, J.S.; Richter, C.; Przybilski, R.; Pitman, A.R.; Fineran, P.C. Cytotoxic Chromosomal Targeting by CRISPR/Cas Systems Can Reshape Bacterial Genomes and Expel or Remodel Pathogenicity Islands. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; He, Y.; Zhang, H.; Liao, X.P.; Liu, Y.H.; Sun, J.; Du, H.; Kreiswirth, B.N.; Chen, L. CRISPR-Cas9-mediated carbapenemase gene and plasmid curing in carbapenem-resistant enterobacteriaceae. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Kim, J.S.; Cho, D.H.; Park, M.; Chung, W.J.; Shin, D.; Ko, K.S.; Kweon, D.H. Crispr/cas9-mediated re-sensitization of antibiotic-resistant escherichia coli harboring extended-spectrum-lactamases. J. Microbiol. Biotechnol. 2015, 26, 394–401. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Y.; Dong, N.; Shen, L.; Zhou, H.; Hu, Y.; Gu, D.; Chen, S.; Zhang, R.; Ji, Q. Application of CRISPR/Cas9-Based Genome Editing in Studying the Mechanism of Pandrug Resistance in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConville, T.H.; Annavajhala, M.K.; Giddins, M.J.; Macesic, N.; Herrera, C.M.; Rozenberg, F.D.; Bhushan, G.L.; Ahn, D.; Mancia, F.; Trent, M.S.; et al. CrrB Positively Regulates High-Level Polymyxin Resistance and Virulence in Klebsiella pneumoniae. Cell Rep. 2020, 33, 8313. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Q.X.; Liu, A.M.; Zhao, G.P.; Wang, J. A Novel and Efficient Method for Bacteria Genome Editing Employing both CRISPR/Cas9 and an Antibiotic Resistance Cassette. Front. Microbiol. 2017, 8, 812. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamel, M.; Rolain, J.-M.; Baron, S.A. The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges. Microorganisms 2021, 9, 442. https://doi.org/10.3390/microorganisms9020442
Hamel M, Rolain J-M, Baron SA. The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges. Microorganisms. 2021; 9(2):442. https://doi.org/10.3390/microorganisms9020442
Chicago/Turabian StyleHamel, Mouna, Jean-Marc Rolain, and Sophie Alexandra Baron. 2021. "The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges" Microorganisms 9, no. 2: 442. https://doi.org/10.3390/microorganisms9020442
APA StyleHamel, M., Rolain, J.-M., & Baron, S. A. (2021). The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges. Microorganisms, 9(2), 442. https://doi.org/10.3390/microorganisms9020442