
Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production



Abstract
:1. Introduction
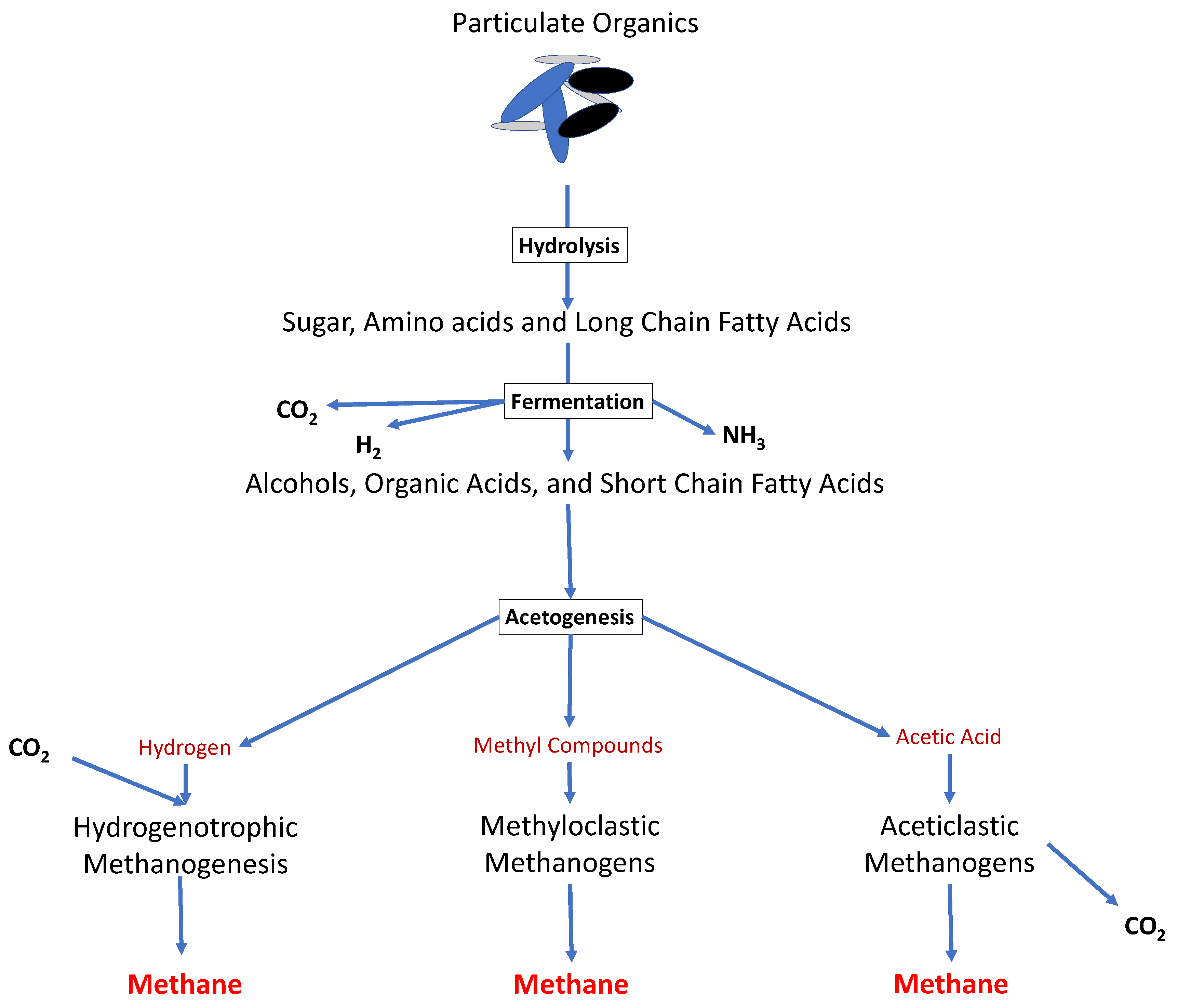
2. Anaerobic Digestion (AD)
3. Microbial Diversity
- the genetics of the organisms and their distribution in the community
- the functional role of their diversity
- the types of species
- the specific amount of each species within the system
4. Determination of Metabolic Functionality by Meta-Omic Techniques
4.1. Metagenomics
4.2. Meta-Transcriptome
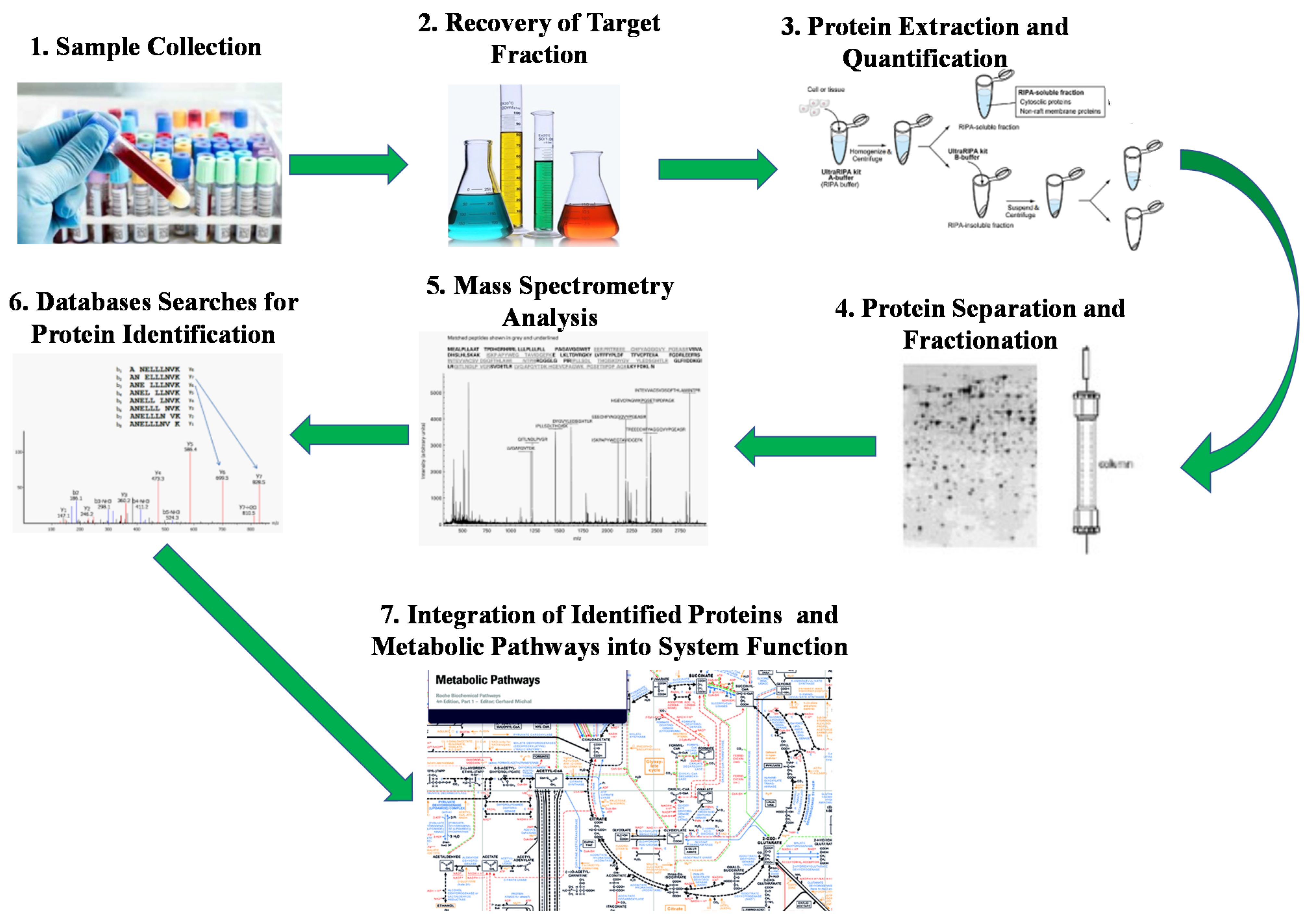
4.3. Meta-Proteome
- contamination by the products of biomass degradation
- sample complexity
- redundant protein identifications
- lack of detailed databases
4.4. Meta-Metabolome
5. Determining Specific Function of Specific Genes and Protein
5.1. Stable Isotope Probing (SIP)
5.2. Fluorescence In Situ Hybridisation (FISH)
5.3. Microautoradiography
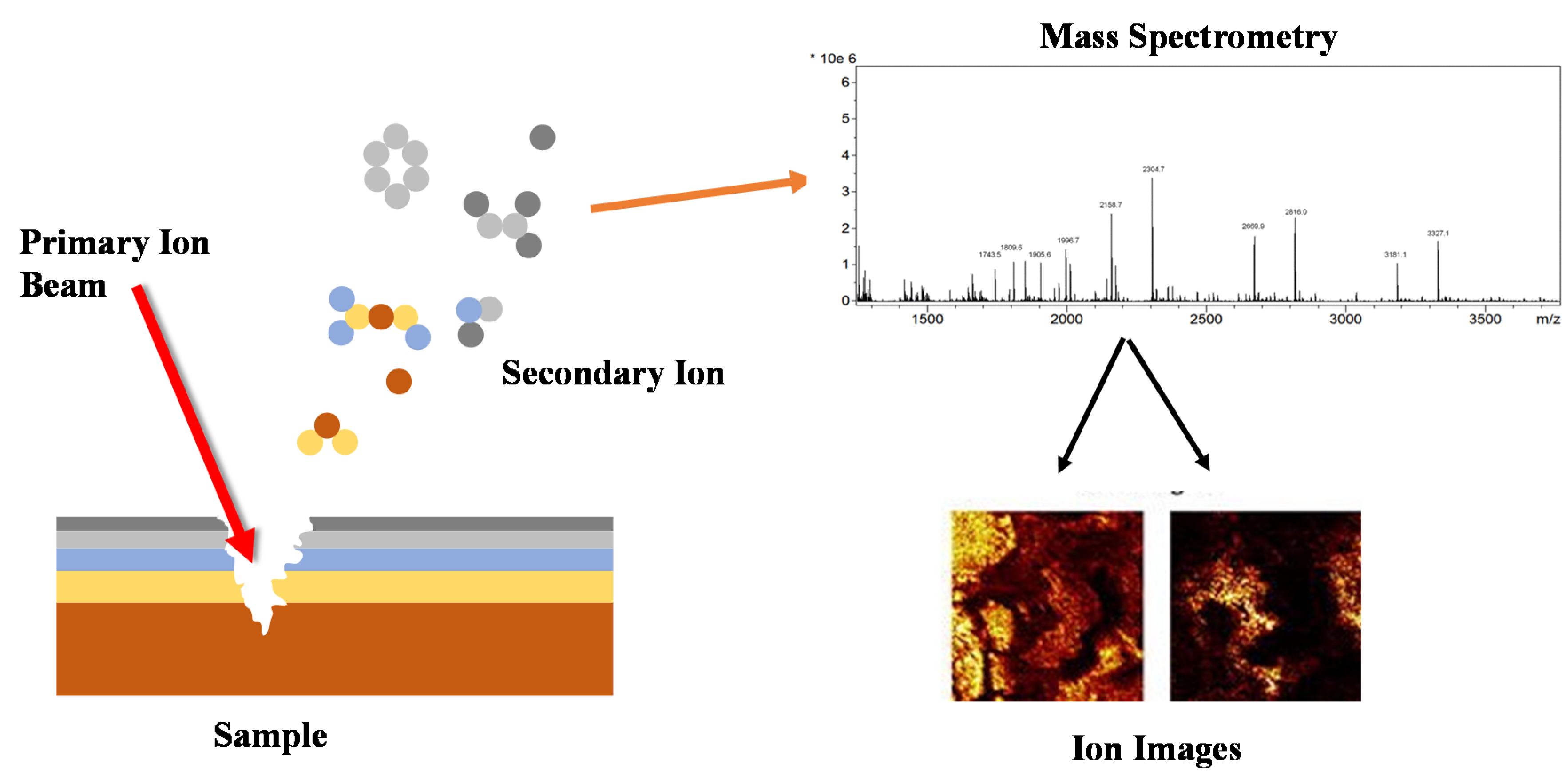
5.4. Secondary Ion Mass Spectroscopy (SIMS)
6. Possible Applications of Molecular Techniques in AD
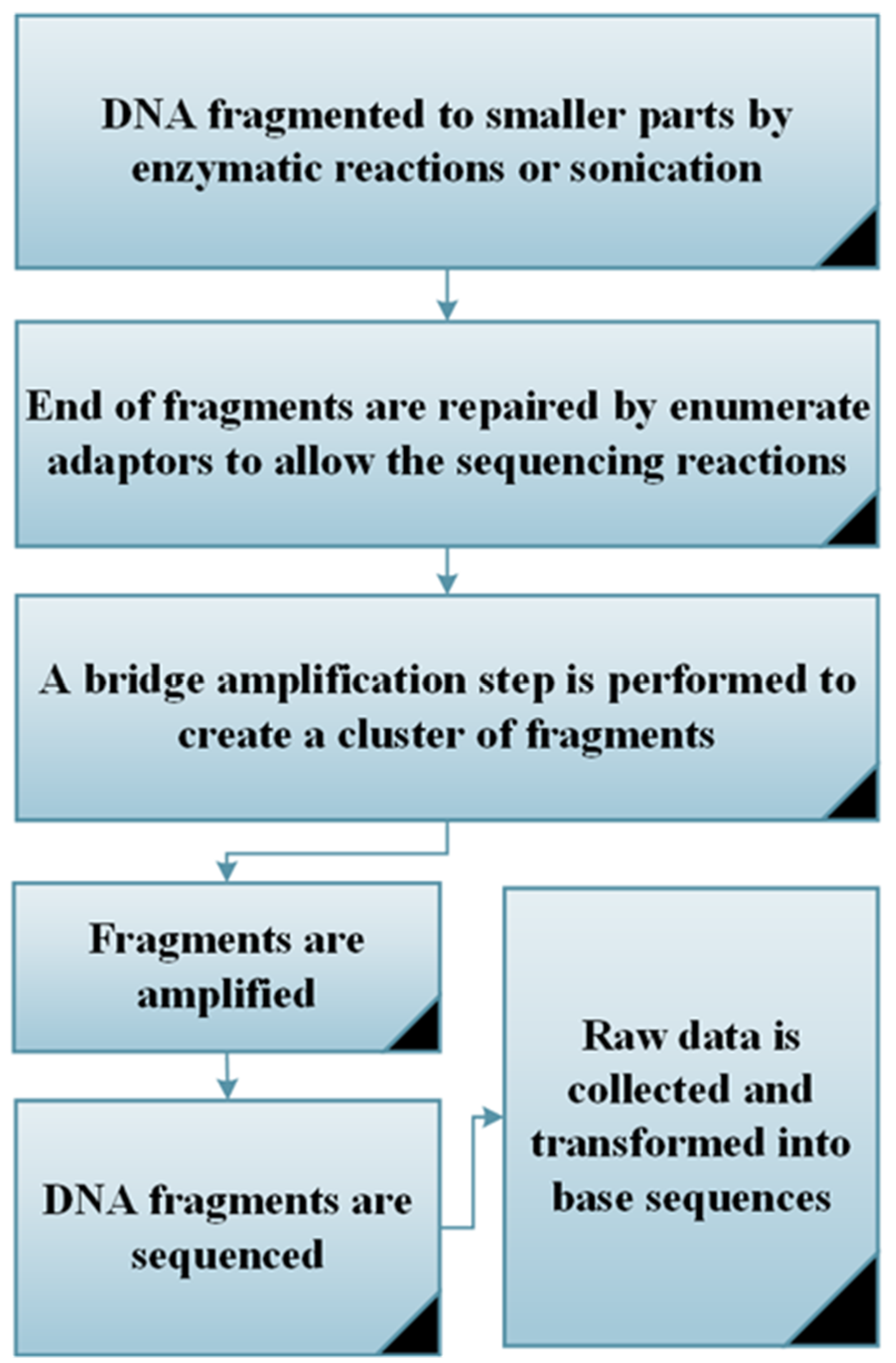
6.1. 16s rRNA Gene Sequencing
6.2. Metagenomics
6.3. Meta-Transcriptome
6.4. Meta-Proteomics
6.5. Meta-Metabolome
7. Application of Microbial Diversity Analysis in AD Models
7.1. Metabolic Models of AD
7.2. Development of Metabolic Models and Main Gaps in the Field
7.3. Examples of Hybrid Cellular-Level Modeling/Biochemical Process Modeling (CLM/BPM) for Enhanced Predictivity of AD Models
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Su, C.; Lei, L.; Duan, Y.; Zhang, K.-Q.; Yang, J. Culture-independent methods for studying environmental microorganisms: Methods, application, and perspective. Appl. Microbiol. Biotechnol. 2012, 93, 993–1003. [Google Scholar] [CrossRef]
- Lamb, J.J. Microbiology of AD. In Anaerobic Digestion: From Biomass to Biogas; SCIO Publishing: Trondheim, Norway, 2020; pp. 53–88. [Google Scholar] [CrossRef]
- Gregorie, E.F.J.; Lamb, J.J.; Lien, K.M.; Pollet, B.G.; Burheim, O.S. Hydrogen and biogas. In Micro-Optics and Energy: Sensors for Energy Devices; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Metcalf, I.; Eddy, G.; Tchobanoglous, H.; Stensel, R.; Tsuchihashi, F. Burton, Wastewater Engineering: Treatment and Resource Recovery, 5th ed.; McGraw-Hill: New York, NY, USA, 2014; Volume 1. [Google Scholar]
- Sarker, S.; Nordgård, A.S.; Lamb, J.J.; Lien, K.M. Biogas and hydrogen. In Hydrogen, Biomass and Bioenergy; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef]
- Zamri, M.; Hasmady, S.; Akhiar, A.; Ideris, F.; Shamsuddin, A.; Mofijur, M.; Fattah, I.M.R.; Mahlia, T. A comprehensive review on anaerobic digestion of organic fraction of municipal solid waste. Renew. Sustain. Energy Rev. 2021, 137, 110637. [Google Scholar] [CrossRef]
- De Vrieze, J.; Pinto, A.J.; Sloan, W.T.; Ijaz, U.Z. The active microbial community more accurately reflects the anaerobic digestion process: 16S rRNA (gene) sequencing as a predictive tool. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Rudakiya, D.M.; Narra, M. Microbial Community Dynamics in Anaerobic Digesters for Biogas Production; Springer: Singapore, 2021; pp. 143–159. [Google Scholar] [CrossRef]
- Lamb, J.J.; Hill, R.E.; Eaton-Rye, J.J.; Hohmann-Marriott, M.F. Functional role of PilA in iron acquisition in the cyanobacterium Synechocystis sp. PCC 6803. PLoS ONE 2014, 9, e105761. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.J.; Hohmann-Marriott, M.F. Manganese acquisition is facilitated by PilA in the cyanobacterium Synechocystis sp. PCC 6803. PLoS ONE 2017, 12, e0184685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanwonterghem, I.; Jensen, P.D.; Ho, D.P.; Batstone, D.J.; Tyson, G.W. Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr. Opin. Biotechnol. 2014, 27, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, M.; Treu, L.; Zhu, X.; Palù, M.; Angelidaki, I.; Campanaro, S.; Kougias, P.G. Microbial dynamics in biogas digesters treating lipid-rich substrates via genome-centric metagenomics. Sci. Total Environ. 2021, 778, 146296. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Morrison, M.; Yu, Z. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour. Technol. 2011, 102, 3730–3739. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Abu Al-Soud, W.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nzila, A.; Razzak, S.A.; Sankara, S.; Nazal, M.K.; Al-Momani, M.; Kang, G.-U.; Ibal, J.C.; Shin, J.-H. Characterisation and microbial community analysis of lipid utilising microorganisms for biogas formation. PLoS ONE 2019, 14, e0224989. [Google Scholar] [CrossRef]
- Sarker, S.; Lamb, J.J.; Hjelme, D.R.; Lien, K.M. A review of the role of critical parameters in the design and operation of biogas production plants. Appl. Sci. 2019, 9, 1915. [Google Scholar] [CrossRef] [Green Version]
- Batstone, D.; Keller, J.; Angelidaki, I.; Kalyuzhnyi, S.; Pavlostathis, S.; Rozzi, A.; Sanders, W.; Siegrist, H.; Vavilin, V. The IWA Anaerobic Digestion Model No 1 (ADM1). Water Sci. Technol. 2002, 45, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, M.R.; Torii, S. Anaerobic co-digestion of cafeteria, vegetable and fruit wastes for biogas production. In 2014 International Conference on Renewable Energy Research and Application (ICRERA), 3rd ed.; Institute of Electrical and Electronics Engineers Inc.: Piscataway, NJ, USA, 2014; pp. 369–374. [Google Scholar] [CrossRef]
- Aryal, N.; Kvist, T.; Ammam, F.; Pant, D.; Ottosen, L.D. An overview of microbial biogas enrichment. Bioresour. Technol. 2018, 264, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Anderson, I.; Ulrich, L.E.; Lupa, B.; Susanti, D.; Porat, I.; Hooper, S.D.; Lykidis, A.; Sieprawska-Lupa, M.; Dharmarajan, L.; Goltsman, E.; et al. Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS ONE 2009, 4, e5797. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.W.; Park, T.; Tong, Y.W.; Yu, Z. The microbiome driving anaerobic digestion and microbial analysis. Adv. Bioenergy 2020, 5, 1–61. [Google Scholar] [CrossRef]
- Feldewert, C.; Lang, K.; Brune, A. The hydrogen threshold of obligately methyl-reducing methanogens. FEMS Microbiol. Lett. 2020, 367, 137. [Google Scholar] [CrossRef]
- Venkiteshwaran, K.; Bocher, B.; Maki, J.; Zitomer, D. Relating Anaerobic Digestion Microbial Community and Process Function. Microbiol. Insights 2015, 8, 37–44. [Google Scholar] [PubMed] [Green Version]
- Esquivel-Elizondo, S.; Parameswaran, P.; Delgado, A.G.; Maldonado, J.; Rittmann, B.E.; Krajmalnik-Brown, R. Archaea and Bacteria Acclimate to High Total Ammonia in a Methanogenic Reactor Treating Swine Waste. Archaea 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Angelidaki, I.; Treu, L.; Tsapekos, P.; Luo, G.; Campanaro, S.; Wenzel, H.; Kougias, P.G. Biogas upgrading and utilization: Current status and perspectives. Biotechnol. Adv. 2018, 36, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christou, M.; Vasileiadis, S.; Kalamaras, S.; Karpouzas, D.; Angelidaki, I.; Kotsopoulos, T. Ammonia-induced inhibition of manure-based continuous biomethanation process under different organic loading rates and associated microbial community dynamics. Bioresour. Technol. 2020, 320, 124323. [Google Scholar] [CrossRef]
- Adekunle, K.F.; Okolie, J.A. A Review of Biochemical Process of Anaerobic Digestion. Adv. Biosci. Biotechnol. 2015, 6, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Hashemi, B.; Sarker, S.; Lamb, J.J.; Lien, K.M. Yield improvements in anaerobic digestion of lignocellulosic feedstocks. J. Clean. Prod. 2021, 288, 125447. [Google Scholar] [CrossRef]
- Ma, J.; Frear, C.; Wang, Z.-W.; Yu, L.; Zhao, Q.; Li, X.; Chen, S. A simple methodology for rate-limiting step determination for anaerobic digestion of complex substrates and effect of microbial community ratio. Bioresour. Technol. 2013, 134, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Nabaterega, R.; Khoei, S.; Eskicioglu, C. Insight into interactions between syntrophic bacteria and archaea in anaerobic digestion amended with conductive materials. Renew. Sustain. Energy Rev. 2021, 144, 110965. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, T.; Si, B.; Watson, J.; Zhang, Y. Accelerating anaerobic digestion for methane production: Potential role of direct interspecies electron transfer. Renew. Sustain. Energy Rev. 2021, 145, 111069. [Google Scholar] [CrossRef]
- Wu, L.-J.; Li, X.-X.; Yang, F.; Zhou, Q.; Ren, R.-P.; Lyu, Y.-K. One-step acquirement of superior microbial communities from mesophilic digested sludge to upgrade anaerobic digestion. Chemosphere 2021, 263, 128047. [Google Scholar] [CrossRef]
- Lamb, J.J.; Bernard, O.; Sarker, S.; Lien, K.M.; Hjelme, D.R. Perspectives of optical colourimetric sensors for anaerobic digestion. Renew. Sustain. Energy Rev. 2019, 111, 87–96. [Google Scholar] [CrossRef]
- Lamb, J.J.; Bernard, O.; Sarker, S.; Lien, K.M.; Hjelme, D.R. Perspectives of surface plasmonic resonance optical fibre sensors for anaerobic digestion. Eng. Life Sci. 2019, 19, 759–769. [Google Scholar] [CrossRef]
- Jain, S.; Jain, S.; Wolf, I.T.; Lee, J.; Tong, Y.W. Tong, A comprehensive review on operating parameters and different pretreatment methodologies for anaerobic digestion of municipal solid waste. Renew. Sustain. Energy Rev. 2015, 52, 142–154. [Google Scholar] [CrossRef]
- Lamb, J.J.; Lien, K.M. Promising selected biohydrogen solutions. In Hydrogen, Biomass and Bioenergy Integration Pathways for Renewable Energy Applications; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Fang, H.H.P.; Zhang, T.; Kamagata, Y. Syntrophy in Anaerobic Digestion. In Anaerobic Biotechnology; Imperial College Press: London, UK, 2015; pp. 13–30. [Google Scholar] [CrossRef]
- Vester, J.K.; Glaring, M.A.; Stougaard, P. Improved cultivation and metagenomics as new tools for bioprospecting in cold environments. Extremophiles 2015, 19, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madigan, M.T.; Martinko, J.M.; Stahl, D.A.; Clark, D.P. Brock Biology of Microorganisms, Global Edition; Pearson: London, UK, 2012. [Google Scholar]
- Cocolin, L.; Alessandria, V.; Dolci, P.; Gorra, R.; Rantsiou, K. Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int. J. Food Microbiol. 2013, 167, 29–43. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Prescott, H.C.; Martinez, F.J.; Curtis, J.L.; Lama, V.N.; Huffnagle, G.B. Analysis of culture-dependent versus culture-independent techniques for identification of bacteria in clinically obtained bronchoalveolar lavage fluid. J. Clin. Microbiol. 2014, 52, 3605–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Mazéas, L.; Sghir, A.; LeBlon, G.; Bouchez, T. Insights into networks of functional microbes catalysing methanization of cellulose under mesophilic conditions. Environ. Microbiol. 2009, 11, 889–904. [Google Scholar] [CrossRef]
- Ito, T.; Yoshiguchi, K.; Ariesyady, H.D.; Okabe, S. Identification and quantification of key microbial trophic groups of methanogenic glucose degradation in an anaerobic digester sludge. Bioresour. Technol. 2012, 123, 599–607. [Google Scholar] [CrossRef]
- Torsvik, V.; Daae, F.L.; Sandaa, R.-A.; Øvreås, L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J. Biotechnol. 1998, 64, 53–62. [Google Scholar] [CrossRef]
- Mirmohamadsadeghi, S.; Karimi, K.; Azarbaijani, R.; Yeganeh, L.P.; Angelidaki, I.; Nizami, A.-S.; Bhat, R.; Dashora, K.; Vijay, V.K.; Aghbashlo, M.; et al. Pretreatment of lignocelluloses for enhanced biogas production: A review on influencing mechanisms and the importance of microbial diversity. Renew. Sustain. Energy Rev. 2021, 135, 110173. [Google Scholar] [CrossRef]
- Fakruddin, M.; Mannan, K.S. Methods for analyzing diversity of microbial communities in natural environments. Ceylon J. Sci. 2013, 42, 19–33. [Google Scholar] [CrossRef]
- Yannarell, A.C.; Triplett, E.W. Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 2005, 71, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ma, T.; Gao, M.; Gao, P.; Cao, M.; Zhu, X.; Li, G. Characterization of microbial diversity and community in water flooding oil reservoirs in China. World J. Microbiol. Biotechnol. 2012, 28, 3039–3052. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Britschgi, T.B.; Moyer, C.L.; Field, K.G. Genetic diversity in Sargasso Sea bacterioplankton. Nature 1990, 345, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Akyol, Ç.; Ince, O.; Ince, B. Crop-based composting of lignocellulosic digestates: Focus on bacterial and fungal diversity. Bioresour. Technol. 2019, 288, 121549. [Google Scholar] [CrossRef]
- Kulski, J.K. Next-Generation Sequencing—An Overview of the History, Tools, and “Omic” Applications. In Next Generation Sequencing—Advances, Applications and Challenges; IntechOpen: London, UK, 2016. [Google Scholar]
- Leclerc, M.; Delgenes, J.-P.; Godon, J.-J. Diversity of the archaeal community in 44 anaerobic digesters as determined by single strand conformation polymorphism analysis and 16S rDNA sequencing. Environ. Microbiol. 2004, 6, 809–819. [Google Scholar] [CrossRef]
- Churko, J.M.; Mantalas, G.L.; Snyder, M.P.; Wu, J.C. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ. Res. 2013, 112, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Bozan, M.; Akyol, Ç.; Ince, O.; Aydin, S.; Ince, B. Application of next-generation sequencing methods for microbial monitoring of anaerobic digestion of lignocellulosic biomass. Appl. Microbiol. Biotechnol. 2017, 101, 6849–6864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Banaszak, J.E.; Parameswaran, P.; Alder, J.; Krajmalnik-Brown, R.; Rittmann, B.E. Focused-Pulsed sludge pre-treatment increases the bacterial diversity and relative abundance of acetoclastic methanogens in a full-scale anaerobic digester. Water Res. 2009, 43, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.J.; Knights, D.; Garcia, M.L.; Scalfone, N.B.; Smith, S.; Yarasheski, K.; Cummings, T.A.; Beers, A.R.; Knight, R.; Angenent, L.T. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc. Natl. Acad. Sci. USA 2011, 108, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Kang, H.-J.; Lee, Y.H.; Lee, T.J.; Han, K.; Choi, Y.; Park, H.-D. Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J. Environ. Monit. 2012, 14, 1893–1905. [Google Scholar] [CrossRef]
- Ronaghi, M.; Karamohamed, S.; Pettersson, B.; Uhlen, M.; Nyrén, P. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 1996, 242, 84–89. [Google Scholar] [CrossRef]
- Ari, Ş.; Arikan, M. Next-generation sequencing: Advantages, disadvantages, and future. In Plant Omics: Trends and Applications; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Li, Z.; Bai, X.; Ruparel, H.; Kim, S.; Turro, N.J.; Ju, J. A photocleavable fluorescent nucleotide for DNA sequencing and analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Dong, M.; Ge, M.; Zhu, L.; Ren, L.; Liu, G.; Mu, R. The History and Advances of Reversible Terminators Used in New Generations of Sequencing Technology. Genom. Proteom. Bioinform. 2013, 11, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Quince, C.; Lanzén, A.; Curtis, T.P.; Davenport, R.J.; Hall, N.; Head, I.M.; Read, L.F.; Sloan, W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 2009, 6, 639–641. [Google Scholar] [CrossRef]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Heydari, M.; Miclotte, G.; Demeester, P.; Van De Peer, Y.; Fostier, J. Evaluation of the impact of Illumina error correction tools on de novo genome assembly. BMC Bioinform. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sheikhizadeh, S.; De Ridder, D. ACE: Accurate correction of errors using K-mer tries. Bioinformatics 2015, 31, 3216–3218. [Google Scholar] [CrossRef] [Green Version]
- Nikolenko, S.I.; Korobeynikov, A.; Alekseyev, M. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, Y.; Wu, X.-L.; Chen, D.; Ma, J.; Hwu, W.-M. BLESS: Bloom filter-based error correction solution for high-throughput sequencing reads. Bioinformatics 2014, 30, 1354–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Rajasekaran, S. EC: An efficient error correction algorithm for short reads. BMC Bioinform. 2015, 16, S2. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; et al. Bioinformatics for NGS-based metagenomics and the application to biogas research. J. Biotechnol. 2017, 261, 10–23. [Google Scholar] [CrossRef]
- Pore, S.D.; Shetty, D.; Arora, P.; Maheshwari, S.; Dhakephalkar, P.K. Metagenome changes in the biogas producing community during anaerobic digestion of rice straw. Bioresour. Technol. 2016, 213, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Sun, D.; Dang, Y.; Feng, X.; Huo, D.; Liu, C.; Zheng, K.; Holmes, D.E. Metagenomic analysis reveals that activated carbon aids anaerobic digestion of raw incineration leachate by promoting direct interspecies electron transfer. Water Res. 2019, 161, 570–580. [Google Scholar] [CrossRef]
- Jaenicke, S.; Ander, C.; Bekel, T.; Bisdorf, R.; Dröge, M.; Gartemann, K.-H.; Jünemann, S.; Kaiser, O.; Krause, L.; Tille, F.; et al. Comparative and joint analysis of two metagenomic datasets from a biogas fermenter obtained by 454-pyrosequencing. PLoS ONE 2011, 6, e14519. [Google Scholar] [CrossRef] [Green Version]
- Rademacher, A.; Zakrzewski, M.; Schlüter, A.; Schönberg, M.; Szczepanowski, R.; Goesmann, A.; Pühler, A.; Klocke, M. Characterization of microbial biofilms in a thermophilic biogas system by high-throughput metagenome sequencing. FEMS Microbiol. Ecol. 2011, 79, 785–799. [Google Scholar] [CrossRef] [Green Version]
- Fontana, A.; Campanaro, S.; Treu, L.; Kougias, P.G.; Cappa, F.; Morelli, L.; Angelidaki, I. Performance and genome-centric metagenomics of thermophilic single and two-stage anaerobic digesters treating cheese wastes. Water Res. 2018, 134, 181–191. [Google Scholar] [CrossRef]
- Reed, D.C.; Algar, C.K.; Huber, J.A.; Dick, G.J. Gene-centric approach to integrating environmental genomics and biogeochemical models. Proc. Natl. Acad. Sci. USA 2014, 111, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Duan, N.; Kougias, P.G.; Campanaro, S.; Treu, L.; Angelidaki, I. Evolution of the microbial community structure in biogas reactors inoculated with seeds from different origin. Sci. Total Environ. 2021, 773, 144981. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Muller, E.E.; Glaab, E.; May, P.; Vlassis, N.; Wilmes, P. Condensing the omics fog of microbial communities. Trends Microbiol. 2013, 21, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.-O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.-G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef]
- Carvalhais, L.C.; Dennis, P.G.; Tyson, G.W.; Schenk, P.M. Application of metatranscriptomics to soil environments. J. Microbiol. Methods 2012, 91, 246–251. [Google Scholar] [CrossRef]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; On behalf of COMBACTE Consortium; Tomasch, J.; Deng, Z.; Jarek, M.; Husemann, P.; Wagner-Döbler, I. Stool metatranscriptomics: A technical guideline for mRNA stabilisation and isolation. BMC Genom. 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, A.J.; Brink, B.G.; Siegel, T.N. Efficient and specific oligo-based depletion of rRNA. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bruno, V.M.; Wang, Z.; Marjani, S.L.; Euskirchen, G.M.; Martin, J.; Sherlock, G.; Snyder, M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010, 20, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, P.; Bond, P.L. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004, 6, 911–920. [Google Scholar] [CrossRef]
- Jia, X.; Xi, B.-D.; Li, M.-X.; Yang, Y.; Wang, Y. Metaproteomics analysis of the functional insights into microbial communities of combined hydrogen and methane production by anaerobic fermentation from reed straw. PLoS ONE 2017, 12, e0183158. [Google Scholar] [CrossRef] [Green Version]
- Siggins, A.; Gunnigle, E.; Abram, F. Exploring mixed microbial community functioning: Recent advances in metaproteomics. FEMS Microbiol. Ecol. 2012, 80, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Heyer, R.; Benndorf, D.; Kohrs, F.; De Vrieze, J.; Boon, N.; Hoffmann, M.; Rapp, E.; Schlüter, A.; Sczyrba, A.; Reichl, U. Proteotyping of biogas plant microbiomes separates biogas plants according to process temperature and reactor type. Biotechnol. Biofuels 2016, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Heyer, R.; Kohrs, F.; Reichl, U.; Benndorf, D. Metaproteomics of complex microbial communities in biogas plants. Microb. Biotechnol. 2015, 8, 749–763. [Google Scholar] [CrossRef]
- Abram, F.; Enright, A.-M.; O’Reilly, J.; Botting, C.; Collins, G.; O’Flaherty, V. A metaproteomic approach gives functional insights into anaerobic digestion. J. Appl. Microbiol. 2011, 110, 1550–1560. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef]
- Heyer, R.; Kohrs, F.; Benndorf, D.; Rapp, E.; Kausmann, R.; Heiermann, M.; Klocke, M.; Reichl, U. Metaproteome analysis of the microbial communities in agricultural biogas plants. N. Biotechnol. 2013, 30, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Ito, K.; Date, Y. Environmental metabolomics with data science for investigating ecosystem homeostasis. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 104, 56–88. [Google Scholar] [CrossRef]
- Callaghan, A.V. Metabolomic investigations of anaerobic hydrocarbon-impacted environments. Curr. Opin. Biotechnol. 2013, 24, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Muhmood, A.; Czekała, W.; Mazurkiewicz, J.; Dach, J.; Dong, R. Untargeted metabolite profiling for screening bioactive compounds in digestate of manure under anaerobic digestion. Water 2019, 11, 2420. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Murrell, J.C. When metagenomics meets stable-isotope probing: Progress and perspectives. Trends Microbiol. 2010, 18, 157–163. [Google Scholar] [CrossRef]
- Deng, Y.; Cui, X.; Dumont, M.G. Identification of active aerobic methanotrophs in plateau wetlands using DNA stable isotope probing. FEMS Microbiol. Lett. 2016, 363, fnw168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziels, R.M.; Sousa, D.Z.; Stensel, H.D.; Beck, D.A.C. DNA-SIP based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies. ISME J. 2017, 12, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Toth, C.R.A.; Berdugo-Clavijo, C.; O’Farrell, C.M.; Jones, G.M.; Sheremet, A.; Dunfield, P.F.; Gieg, L.M. Stable Isotope and Metagenomic Profiling of a Methanogenic Naphthalene-Degrading Enrichment Culture. Microorganisms 2018, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Montero, B.; García-Morales, J.; Sales, D.; Solera, R. Analysis of methanogenic activity in a thermophilic-dry anaerobic reactor: Use of fluorescent in situ hybridization. Waste Manag. 2009, 29, 1144–1151. [Google Scholar] [CrossRef]
- Crocetti, G.; Murto, M.; Björnsson, L. An update and optimisation of oligonucleotide probes targeting methanogenic Archaea for use in fluorescence in situ hybridisation (FISH). J. Microbiol. Methods 2006, 65, 194–201. [Google Scholar] [CrossRef]
- Raskin, L.; Stromley, J.M.; Rittmann, B.E.; Stahl, D.A. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 1994, 60, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Raskin, L.; Poulsen, L.K.; Noguera, D.; Rittmann, B.E.; Stahl, D.A. Quantification of methanogenic groups in anaerobic biological reactors by oligonucleotide probe hybridization. Appl. Environ. Microbiol. 1994, 60, 1241–1248. [Google Scholar] [CrossRef] [Green Version]
- Gagliano, M.C.; Sudmalis, D.; Temmink, H.; Plugge, C.M. Calcium effect on microbial activity and biomass aggregation during anaerobic digestion at high salinity. N. Biotechnol. 2020, 56, 114–122. [Google Scholar] [CrossRef]
- Lukumbuzya, M.; Kristensen, J.M.; Kitzinger, K.; Pommerening-Röser, A.; Nielsen, P.H.; Wagner, M.; Daims, H.; Pjevac, P. A refined set of rRNA-targeted oligonucleotide probes for in situ detection and quantification of ammonia-oxidizing bacteria. Water Res. 2020, 186, 116372. [Google Scholar] [CrossRef]
- Sanz, J.L.; Köchling, T. Molecular biology techniques used in wastewater treatment: An overview. Process. Biochem. 2007, 42, 119–133. [Google Scholar] [CrossRef]
- Andreasen, K.; Nielsen, P.H. Application of microautoradiography to the study of substrate uptake by filamentous microorganisms in activated sludge. Appl. Environ. Microbiol. 1997, 63, 3662–3668. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Nielsen, P.H.; Loy, A.; Nielsen, J.L.; Daims, H. Linking microbial community structure with function: Fluorescence in situ hybridization-microautoradiography and isotope arrays. Curr. Opin. Biotechnol. 2006, 17, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Carman, K.R. Radioactive labeling of a natural assemblage of marine sedimentary bacteria and microalgae for trophic studies: An autoradiographic study. Microb. Ecol. 1990, 19, 279–290. [Google Scholar] [CrossRef]
- Ito, T.; Yoshiguchi, K.; Ariesyady, H.D.; Okabe, S. Identification of a novel acetate-utilizing bacterium belonging to Synergistes group 4 in anaerobic digester sludge. ISME J. 2011, 5, 1844–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M. Single-Cell Ecophysiology of Microbes as Revealed by Raman Microspectroscopy or Secondary Ion Mass Spectrometry Imaging. Annu. Rev. Microbiol. 2009, 63, 411–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nürnberg, D.J.; Mariscal, V.; Bornikoel, A.J.; Nieves-Morión, M.; Krauß, N.; Herrero, A.; Maldener, I.; Flores, E.; Mullineaux, C.W. Intercellular diffusion of a fluorescent sucrose analog via the septal junctions in a filamentous cyanobacterium. mBio 2015, 6, e02109-14. [Google Scholar] [CrossRef] [Green Version]
- Popa, R.; Weber, P.K.; Pett-Ridge, J.; Finzi, J.A.; Fallon, S.J.; Hutcheon, I.D.; Nealson, K.H.; Capone, D.G. Carbon and nitrogen fixation and metabolite exchange in and between individual cells of Anabaena oscillarioides. ISME J. 2007, 1, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Pett-Ridge, J.; Weber, P.K. NanoSIP: NanoSIMS applications for microbial biology. Methods Mol. Biol. 2012, 881, 375–408. [Google Scholar] [CrossRef]
- Ho, D.P.; Jensen, P.D.; Batstone, D.J. Methanosarcinaceae and acetate-oxidizing pathways dominate in high-rate thermophilic anaerobic digestion of waste-activated sludge. Appl. Environ. Microbiol. 2013, 79, 6491–6500. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.J.; Garcia, M.L.; Perkins, S.D.; Yarasheski, K.E.; Smith, S.R.; Muegge, B.D.; Stadermann, F.J.; DeRito, C.M.; Floss, C.; Madsen, E.L.; et al. Microbial community dynamics and stability during an ammonia-induced shift to syntrophic acetate oxidation. Appl. Environ. Microbiol. 2014, 80, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhu, X.; Yan, Q.; Zhang, Y.; Angelidaki, I. Microbial community response to ammonia levels in hydrogen assisted biogas production and upgrading process. Bioresour. Technol. 2020, 296, 122276. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Nordgård, A.S.R.; Bergland, W.H.; Vadstein, O.; Mironov, V.; Bakke, R.; Østgaard, K.; Bakke, I. Anaerobic digestion of pig manure supernatant at high ammonia concentrations characterized by high abundances of Methanosaeta and non-euryarchaeotal archaea. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Amha, Y.M.; Anwar, M.Z.; Brower, A.; Jacobsen, C.S.; Stadler, L.B.; Webster, T.M.; Smith, A.L. Inhibition of anaerobic digestion processes: Applications of molecular tools. Bioresour. Technol. 2018, 247, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, E.; Overmann, J. Ecological Significance of Microdiversity: Identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl. Environ. Microbiol. 2004, 70, 4831–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterkamp, M.J.; Bauer, S.; Ibáñez, A.B.; Méndez-García, C.; Hong, P.-Y.; Cann, I.; Mackie, R.I. Identification of methanogenesis and syntrophy as important microbial metabolic processes for optimal thermophilic anaerobic digestion of energy cane thin stillage. Bioresour. Technol. Rep. 2019, 7, 100254. [Google Scholar] [CrossRef]
- Maroniche, G.A.; García, J.E.; Salcedo, F.; Creus, C.M. Molecular identification of Azospirillum spp.: Limitations of 16S rRNA and qualities of rpoD as genetic markers. Microbiol. Res. 2017, 195, 1–10. [Google Scholar] [CrossRef]
- Drake, H.L. Acetogenesis, Acetogenic Bacteria, and the Acetyl-CoA “Wood/Ljungdahl” Pathway: Past and Current Perspectives. In Acetogenesis; Springer: Boston, MA, USA, 1994. [Google Scholar]
- Lamb, J.J.; Hillestad, M.; Rytter, E.; Bock, R.; Nordgård, A.S.R.; Lien, K.M.; Burheim, O.S.; Pollet, B. Traditional routes for hydrogen production and carbon conversion. In Hydrogen, Biomass and Bioenergy: Integration Pathways for Renewable Energy Applications; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Wirth, R.; Kovács, E.; Maróti, G.; Bagi, Z.; Rákhely, G.; Kovács, K.L. Characterization of a biogas-producing microbial community by short-read next generation DNA sequencing. Biotechnol. Biofuels 2012, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.J. Characterization of Bacterial Electron Transport to Extracellular Electron Acceptors. Ph.D. Thesis, Norwegian University of Science and Technology, Trondheim, Norway, 2016; p. 186. [Google Scholar]
- Tyson, G.W.; Lo, I.; Baker, B.J.; Allen, E.E.; Hugenholtz, P.; Banfield, J.F. Genome-directed isolation of the key nitrogen fixer leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl. Environ. Microbiol. 2005, 71, 6319–6324. [Google Scholar] [CrossRef] [Green Version]
- Pope, P.; Smith, W.; Denman, S.E.; Tringe, S.G.; Barry, K.; Hugenholtz, P.; McSweeney, C.S.; McHardy, A.C.; Morrison, M. Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar wallabies. Science 2011, 333, 646–648. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Smith, D.P.; Gilbert, J.A. Beyond the genome: Community-level analysis of the microbial world. Biol. Philos. 2012, 28, 261–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Loh, K.-C.; Lim, J.W.; Zhang, J. Bioinformatics analysis of metagenomics data of biogas-producing microbial communities in anaerobic digesters: A review. Renew. Sustain. Energy Rev. 2019, 100, 110–126. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Goesmann, A.; Jaenicke, S.; Jünemann, S.; Eikmeyer, F.; Szczepanowski, R.; Abu Al-Soud, W.; Sørensen, S.; Pühler, A.; Schlüter, A. Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J. Biotechnol. 2012, 158, 248–258. [Google Scholar] [CrossRef]
- Jia, Y.; Ng, S.-K.; Lu, H.; Cai, M.; Lee, P.K.H. Genome-centric metatranscriptomes and ecological roles of the active microbial populations during cellulosic biomass anaerobic digestion. Biotechnol. Biofuels 2018, 11, 117. [Google Scholar] [CrossRef]
- Nolla-Ardèvol, V.; Strous, M.; Tegetmeyer, H.E. Anaerobic digestion of the microalga Spirulina at extreme alkaline conditions: Biogas production, metagenome, and metatranscriptome. Front. Microbiol. 2015, 6, 597. [Google Scholar] [CrossRef]
- Morita, M.; Malvankar, N.S.; Franks, A.; Summers, Z.M.; Giloteaux, L.; Rotaru, A.-E.; Rotaru, C.; Lovley, D.R. Potential for direct interspecies electron transfer in methanogenic wastewater digester aggregates. mBio 2011, 2, e00159-11. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Rotaru, A.-E.; Shrestha, P.M.; Malvankar, N.S.; Nevin, K.P.; Lovley, D.R. Promoting direct interspecies electron transfer with activated carbon. Energy Environ. Sci. 2012, 5, 8982–8989. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, P.M.; Rotaru, A.-E.; Summers, Z.M.; Shrestha, M.; Liu, F.; Lovley, D.R. Transcriptomic and genetic analysis of direct interspecies electron transfer. Appl. Environ. Microbiol. 2013, 79, 2397–2404. [Google Scholar] [CrossRef] [Green Version]
- Winkler, M.K.H.; Kleerebezem, R.; Kuenen, J.G.; Yang, J.; Van Loosdrecht, M.C.M. Segregation of biomass in cyclic anaerobic/aerobic granular sludge allows the enrichment of anaerobic ammonium oxidizing bacteria at low temperatures. Environ. Sci. Technol. 2011, 45, 7330–7337. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Kougias, P.G.; Treu, L.; Kovalovszki, A.; Valle, G.; Cappa, F.; Morelli, L.; Angelidaki, I.; Campanaro, S. Microbial activity response to hydrogen injection in thermophilic anaerobic digesters revealed by genome-centric metatranscriptomics. Microbiome 2018, 6, 194. [Google Scholar] [CrossRef]
- Grohmann, A.; Fehrmann, S.; Vainshtein, Y.; Haag, N.L.; Wiese, F.; Stevens, P.; Naegele, H.-J.; Oechsner, H.; Hartsch, T.; Sohn, K.; et al. Microbiome dynamics and adaptation of expression signatures during methane production failure and process recovery. Bioresour. Technol. 2018, 247, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.R.; Dwyer, J.; Drozdov, I.; Yin, X.; Mayr, M. Proteomics: From single molecules to biological pathways. Cardiovasc. Res. 2012, 97, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.; Benndorf, D.; Rapp, E.; Reichl, U.; Palese, L.L.; Pollice, A. Metaproteome analysis of sewage sludge from membrane bioreactors. Proteomics 2011, 11, 2738–2744. [Google Scholar] [CrossRef]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schlüter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Pühler, A.; Reichl, U.; Klocke, M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst. Appl. Microbiol. 2013, 36, 330–338. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Viant, M.; Tjeerdema, R.S. Metabolomics: Methodologies and applications in the environmental sciences. J. Pestic. Sci. 2006, 31, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Scaglia, B.; Pognani, M.; Adani, F. Evaluation of hormone-like activity of the dissolved organic matter fraction (DOM) of compost and digestate. Sci. Total Environ. 2015, 514, 314–321. [Google Scholar] [CrossRef]
- Angelidaki, I.; Karakashev, D.; Batstone, D.J.; Plugge, C.M.; Stams, A.J. Biomethanation and its potential. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2011; pp. 327–351. [Google Scholar]
- Lyberatos, G.; Skiadas, I.V. Modeling of Anaerobic Digestion—A Review. Glob. NEST Int. J. 1999, 1, 63–76. [Google Scholar] [CrossRef]
- Batstone, D.; Hülsen, T.; Oehmen, A. Metabolic modelling of mixed culture anaerobic microbial processes. Curr. Opin. Biotechnol. 2019, 57, 137–144. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, H.U.; Lee, S.Y. Current state and applications of microbial genome-scale metabolic models. Curr. Opin. Syst. Biol. 2017, 2, 10–18. [Google Scholar] [CrossRef]
- González-Cabaleiro, R.; Lema, J.M.; Rodríguez, J. Metabolic energy-based modelling explains product yielding in anaerobic mixed culture fermentations. PLoS ONE 2015, 10, e0126739. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Sohn, S.B.; Bin Kim, Y.; Kim, W.J.; Lee, S.Y. Recent advances in reconstruction and applications of genome-scale metabolic models. Curr. Opin. Biotechnol. 2012, 23, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Thor, S.; Peterson, J.R.; Luthey-Schulten, Z. Genome-Scale Metabolic Modeling of Archaea Lends Insight into Diversity of Metabolic Function. Archaea 2017, 2017, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelidaki, I.; Ellegaard, L.; Ahring, B.K. A comprehensive model of anaerobic bioconversion of complex substrates to biogas. Biotechnol. Bioeng. 1999, 63, 363–372. [Google Scholar] [CrossRef]
- Henze, M.; Gujer, W.; Mino, T.; Van Loosedrecht, M. Activated Sludge Models ASM1, ASM2, ASM2d and ASM3; IWA Publishing: London, UK, 2006; Volume 5. [Google Scholar] [CrossRef]
- Cai, Y.; Zheng, Z.; Wang, X. Obstacles faced by methanogenic archaea originating from substrate-driven toxicants in anaerobic digestion. J. Hazard. Mater. 2021, 403, 123938. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnyi, S.; Fedorovich, V. Mathematical modelling of competition between sulphate reduction and methanogenesis in anaerobic reactors. Bioresour. Technol. 1998, 65, 227–242. [Google Scholar] [CrossRef]
- Regueira, A.; González-Cabaleiro, R.; Ofiţeru, I.; Rodríguez, J.; Lema, J. Electron bifurcation mechanism and homoacetogenesis explain products yields in mixed culture anaerobic fermentations. Water Res. 2018, 141, 349–356. [Google Scholar] [CrossRef]
- Mosey, F.E. Mathematical modelling of the anaerobic digestion process: Regulatory mechanisms for the formation of short-chain volatile acids from glucose. Water Sci. Technol. 1983, 15, 209–232. [Google Scholar] [CrossRef]
- Hoelzle, R.D.; Virdis, B.; Batstone, D.J. Regulation mechanisms in mixed and pure culture microbial fermentation. Biotechnol. Bioeng. 2014, 111, 2139–2154. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, I.R.; Pullammanappallil, P.C. Protein degradation during anaerobic wastewater treatment: Derivation of stoichiometry. Biodegradation 2001, 12, 247–256. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Y.; Liu, S.; Lévesque, C.L.; Zhao, F.; Yin, J.; Dong, B. Branched chain amino acids alter fatty acid profile in colostrum of sows fed a high fat diet. J. Anim. Sci. Biotechnol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Roman-Garcia, Y.; Denton, B.; Mitchell, K.; Lee, C.; Socha, M.; Firkins, J. Conditions stimulating neutral detergent fiber degradation by dosing branched-chain volatile fatty acids. I: Comparison with branched-chain amino acids and forage source in ruminal batch cultures. J. Dairy Sci. 2021, 104, 6739–6755. [Google Scholar] [CrossRef] [PubMed]
- Stams, A.J.M.; Plugge, C.M. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Genet. 2009, 7, 568–577. [Google Scholar] [CrossRef]
- Boone, D.R.; Johnson, R.L.; Liu, Y. Diffusion of the Interspecies Electron Carriers H2 and Formate in Methanogenic Ecosystems and Its Implications in the Measurement of Km for H2 or Formate Uptake. Appl. Environ. Microbiol. 1989, 55, 1735–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Embree, M.; Liu, J.K.; Al-Bassam, M.M.; Zengler, K. Networks of energetic and metabolic interactions define dynamics in microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 15450–15455. [Google Scholar] [CrossRef] [Green Version]
- Westerholm, M.; Isaksson, S.; Lindsjö, O.K.; Schnürer, A. Microbial community adaptability to altered temperature conditions determines the potential for process optimisation in biogas production. Appl. Energy 2018, 226, 838–848. [Google Scholar] [CrossRef]
- Rotaru, A.-E.; Calabrese, F.; Stryhanyuk, H.; Musat, F.; Shrestha, P.M.; Weber, H.S.; Snoeyenbos-West, O.L.O.; Hall, P.O.J.; Richnow, H.H.; Musat, N.; et al. Conductive particles enable syntrophic acetate oxidation between Geobacter and methanosarcina from coastal sediments. mBio 2018, 9, e00226-18. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.; Rodríguez, J. Modelling sulfate reduction in anaerobic digestion: Complexity evaluation and parameter calibration. Water Res. 2018, 130, 255–262. [Google Scholar] [CrossRef]
- TStorck, T.; Virdis, B.; Batstone, D.J. Modelling extracellular limitations for mediated versus direct interspecies electron transfer. ISME J. 2016, 10, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, I.; Volcke, E.I.; Rajinikanth, R.; Steyer, J.-P. Modeling microbial diversity in anaerobic digestion through an extended ADM1 model. Water Res. 2009, 43, 2787–2800. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, S.; Koch, S.; Bonk, F.; Popp, D.; Benndorf, D.; Klamt, S.; Centler, F. Augmenting biogas process modeling by resolving intracellular metabolic activity. Front. Microbiol. 2019, 10, 1095. [Google Scholar] [CrossRef]
- Jimenez, J.; Charnier, C.; Kouas, M.; Latrille, E.; Torrijos, M.; Harmand, J.; Patureau, D.; Sperandio, M.; Morgenroth, E.; Béline, F.; et al. Modelling hydrolysis: Simultaneous versus sequential biodegradation of the hydrolysable fractions. Waste Manag. 2020, 101, 150–160. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Name | Reaction | ∆G° (kJ/mol) |
|---|---|---|
| Syntrophic Acetate Oxidation | CH3COO− + 4H2O → 2HCO3− + 4H2 + H+ | +104.6 |
| Hydrogen to Methane | 4H2 + HCO3− + H+ → CH4 + 3H2O | −135.6 |
| Overall Reaction | CH3COO− + H2O → CH4 + HCO3− | −31 |
| Molecular Biotechniques | Biochemical Techniques |
|---|---|
| Mole percentage guanine-cytosine Nucleic acid hybridization DNA reassociation, restriction fragment length polymorphism (RFLP) Terminal restriction fragment length polymorphism (T-RFLP) Ribosomal intergenic spacer analysis (RISA) Automated ribosomal intergenic spacer analysis (ARISA) Amplified ribosomal DNA restriction analysis (ARDRA) DNA microarrays Polymerase chain reaction (PCR) | Plate counts Sole-carbon-source utilization (SCSU) Phospholipid fatty acid (PLFA) analysis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashemi, S.; Hashemi, S.E.; Lien, K.M.; Lamb, J.J. Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production. Microorganisms 2021, 9, 1162. https://doi.org/10.3390/microorganisms9061162
Hashemi S, Hashemi SE, Lien KM, Lamb JJ. Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production. Microorganisms. 2021; 9(6):1162. https://doi.org/10.3390/microorganisms9061162
Chicago/Turabian StyleHashemi, Seyedbehnam, Sayed Ebrahim Hashemi, Kristian M. Lien, and Jacob J. Lamb. 2021. "Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production" Microorganisms 9, no. 6: 1162. https://doi.org/10.3390/microorganisms9061162
APA StyleHashemi, S., Hashemi, S. E., Lien, K. M., & Lamb, J. J. (2021). Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production. Microorganisms, 9(6), 1162. https://doi.org/10.3390/microorganisms9061162