
Full-Length SSU rRNA Gene Sequencing Allows Species-Level Detection of Bacteria, Archaea, and Yeasts Present in Milk

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Milk Samples
2.2. DNA Extraction of the Bovine Milk
2.3. DNA Extraction of the Mock Communities
2.4. Short Amplicon 16S rRNA Gene Library Preparation
2.5. Library Quality Check and the Sequencing of the Short Amplicons
2.6. Synthetic Long-Read Sequencing Using the LoopSeq 16S & 18S Microbiome Kit
2.7. Data Analysis of the Short Reads Using DADA2
3. Results
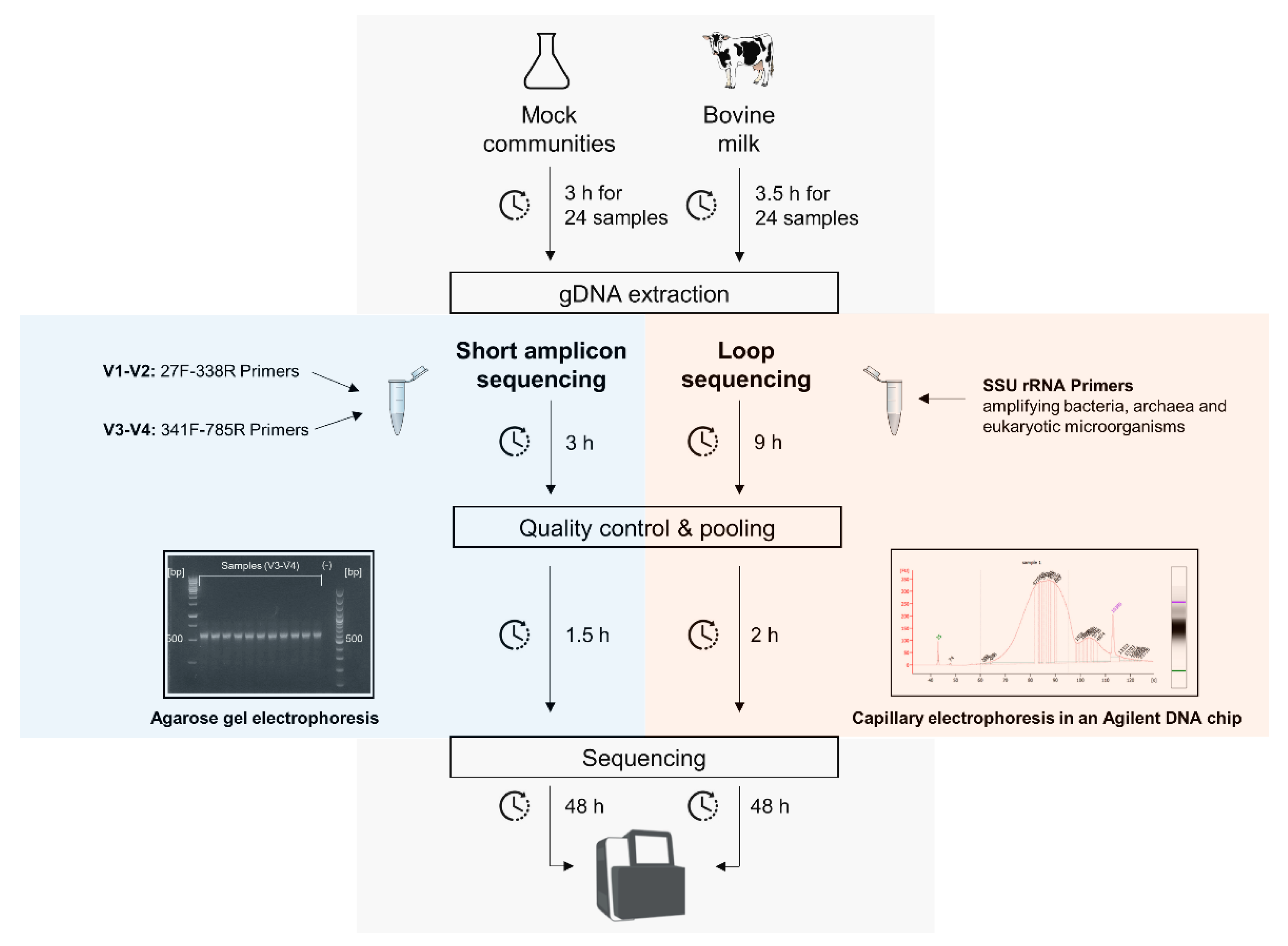
3.1. Protocol Overview for the Short and Full-Length 16S rRNA Gene Sequencing
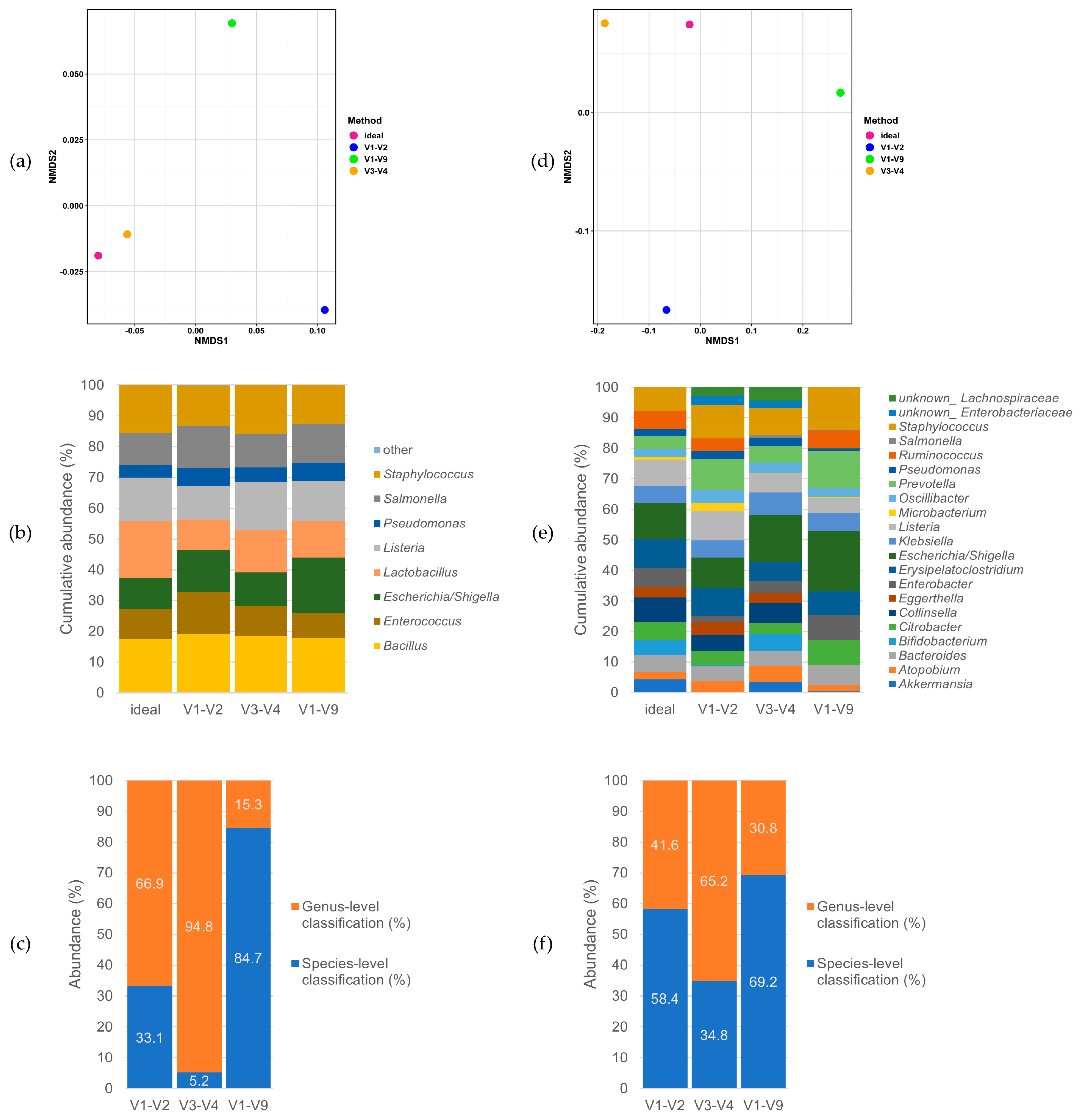
3.2. Performance on Mock Communities
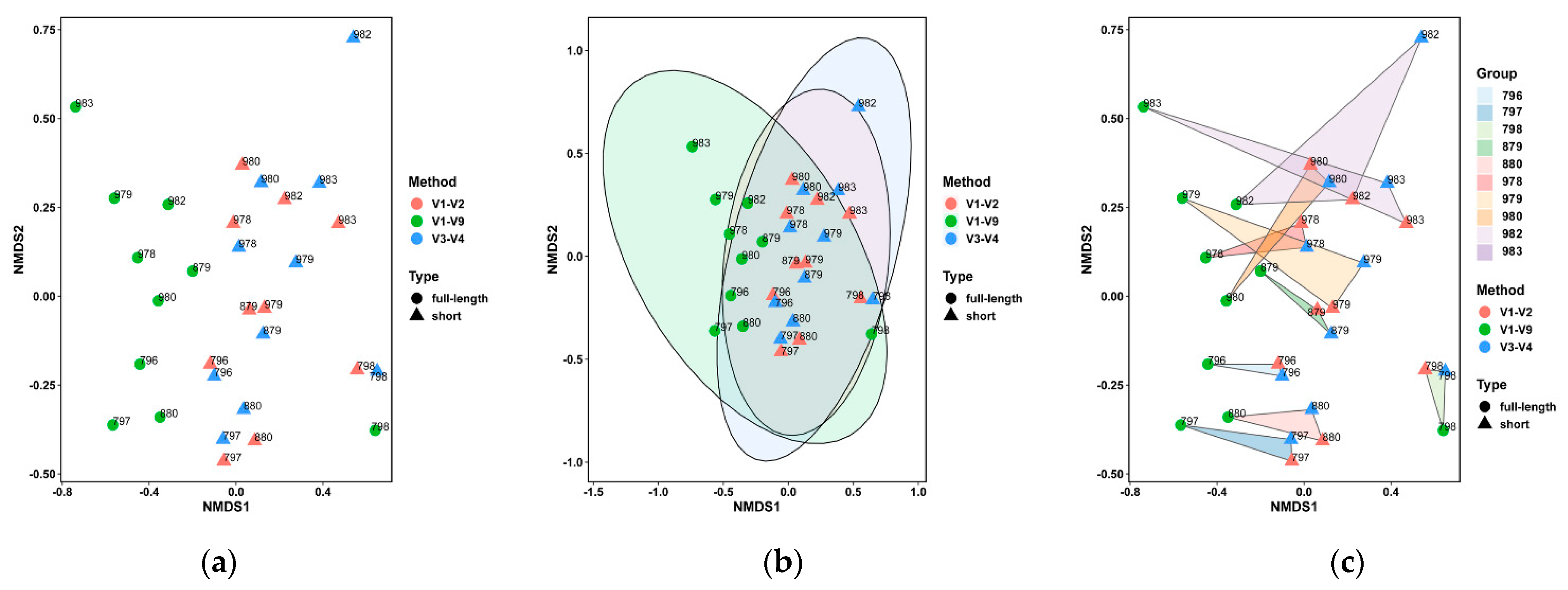
3.3. Performance on the Bovine Milk Samples for Bacteria Identification
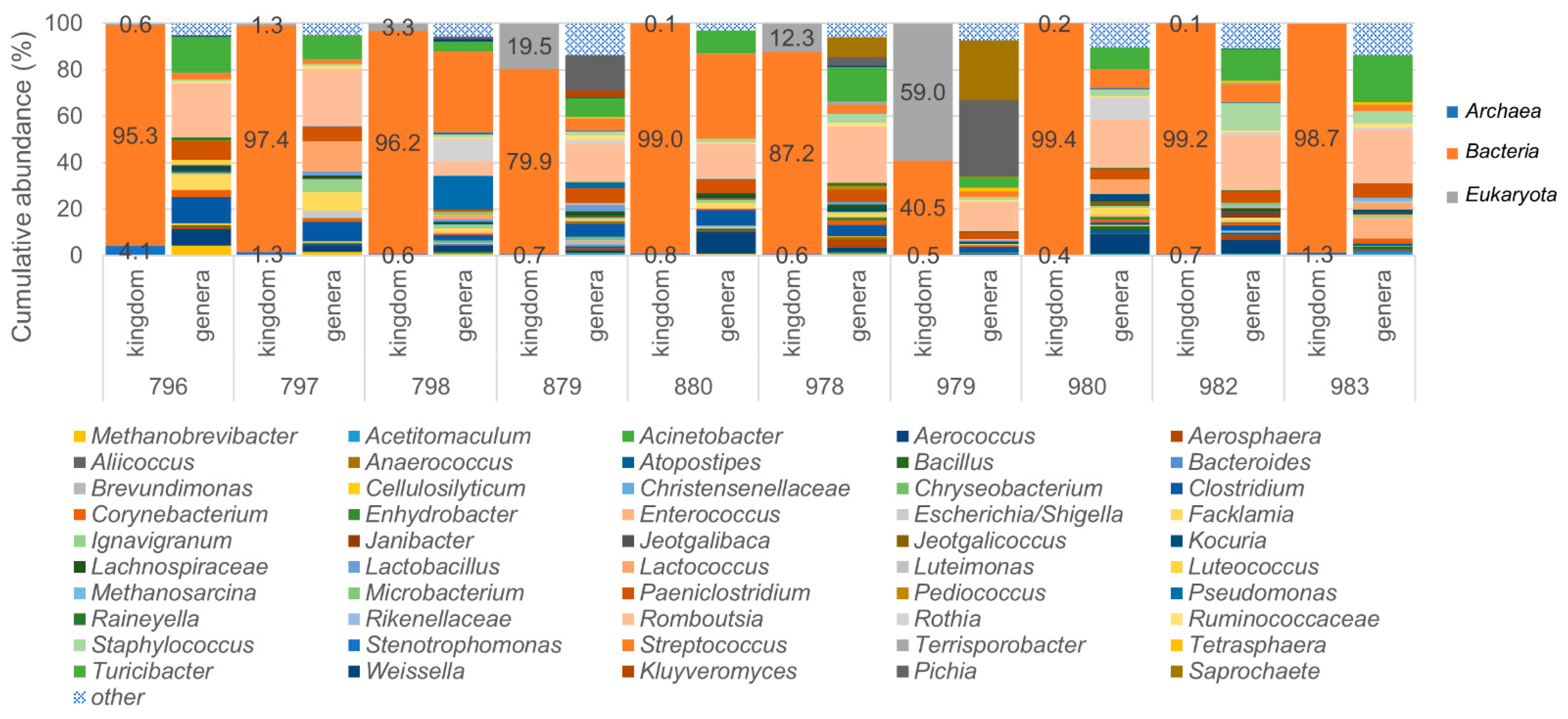
3.4. Identification of Archaea and Eukaryotes in Bovine Milk Samples Using the LoopSeq Protocol
3.5. Identification of Putative Mastitis-Causing Pathogens
4. Discussion
4.1. Full-Length SSU rRNA Gene Sequencing Improves Species-Level Classification but Shows Primer Issues
4.2. Using Full-Length Sequencing Approaches for Microbial Monitoring
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.B. 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. Mol. Diagn. 2001, 6, 313–321. [Google Scholar] [CrossRef]
- Reitmeier, S.; Kiessling, S.; Neuhaus, K.; Haller, D. Comparing Circadian Rhythmicity in the Human Gut Microbiome. STAR Protoc. 2020, 100148. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.M.; Darling, A.E. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq. PeerJ 2016, 4, e2492. [Google Scholar] [CrossRef]
- Karst, S.M.; Dueholm, M.S.; McIlroy, S.J.; Kirkegaard, R.H.; Nielsen, P.H.; Albertsen, M. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat. Biotechnol. 2018, 36, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M. Prokaryotic DNA Transposons: Classes and Mechanism. eLS 2017, 1–16. [Google Scholar] [CrossRef]
- Porcellato, D.; Meisal, R.; Bombelli, A.; Narvhus, J.A. A core microbiota dominates a rich microbial diversity in the bovine udder and may indicate presence of dysbiosis. Sci. Rep. 2020, 10, 21608. [Google Scholar] [CrossRef] [PubMed]
- Taponen, S.; McGuinness, D.; Hiitiö, H.; Simojoki, H.; Zadoks, R.; Pyörälä, S. Bovine milk microbiome: A more complex issue than expected. Vet. Res. 2019, 50, 44. [Google Scholar] [CrossRef] [Green Version]
- Metzger, S.A.; Hernandez, L.L.; Skarlupka, J.H.; Walker, T.M.; Suen, G.; Ruegg, P.L. A Cohort Study of the Milk Microbiota of Healthy and Inflamed Bovine Mammary Glands From Dryoff Through 150 Days in Milk. Front. Vet. Sci. 2018, 5, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremonesi, P.; Ceccarani, C.; Curone, G.; Severgnini, M.; Pollera, C.; Bronzo, V.; Riva, F.; Addis, M.F.; Filipe, J.; Amadori, M.; et al. Milk microbiome diversity and bacterial group prevalence in a comparison between healthy Holstein Friesian and Rendena cows. PLoS ONE 2018, 13, e0205054. [Google Scholar] [CrossRef] [Green Version]
- Metzger, S.A.; Hernandez, L.L.; Skarlupka, J.H.; Suen, G.; Walker, T.M.; Ruegg, P.L. Influence of sampling technique and bedding type on the milk microbiota: Results of a pilot study. J. Dairy Sci. 2018, 101, 6346–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, M.; Xie, X.; Bao, H.; Sun, L.; He, T.; Zhao, H.; Zhou, Y.; Zhang, L.; Zhang, H.; Wei, R.; et al. Insights Into the Bovine Milk Microbiota in Dairy Farms With Different Incidence Rates of Subclinical Mastitis. Front. Microbiol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Doyle, C.J.; Gleeson, D.; O’Toole, P.W.; Cotter, P.D. High-throughput metataxonomic characterization of the raw milk microbiota identifies changes reflecting lactation stage and storage conditions. Int. J. Food Microbiol. 2017, 255, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Oultram, J.W.H.; Ganda, E.K.; Boulding, S.C.; Bicalho, R.C.; Oikonomou, G. A Metataxonomic Approach Could Be Considered for Cattle Clinical Mastitis Diagnostics. Front. Vet. Sci. 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Sokolov, S.; Fursova, K.; Shulcheva, I.; Nikanova, D.; Artyemieva, O.; Kolodina, E.; Sorokin, A.; Dzhelyadin, T.; Shchannikova, M.; Shepelyakovskaya, A.; et al. Comparative Analysis of Milk Microbiomes and Their Association with Bovine Mastitis in Two Farms in Central Russia. Animals 2021, 11, 1401. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; You, C.; Ren, J.; Chen, W.; Zheng, H.; Liu, Z. Variation in Raw Milk Microbiota Throughout 12 Months and the Impact of Weather Conditions. Sci. Rep. 2018, 8, 2371. [Google Scholar] [CrossRef] [Green Version]
- Catozzi, C.; Ceciliani, F.; Lecchi, C.; Talenti, A.; Vecchio, D.; De Carlo, E.; Grassi, C.; Sánchez, A.; Francino, O.; Cuscó, A. Short communication: Milk microbiota profiling on water buffalo with full-length 16S rRNA using nanopore sequencing. J. Dairy Sci. 2020, 103, 2693–2700. [Google Scholar] [CrossRef]
- Contreras, G.A.; Rodríguez, J.M. Mastitis: Comparative Etiology and Epidemiology. J. Mammary Gland Biol. Neoplasia 2011, 16, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Dufour, S.; Labrie, J.; Jacques, M. The Mastitis Pathogens Culture Collection. Microbiol. Resour. Announc. 2019, 8, e00133-19. [Google Scholar] [CrossRef] [Green Version]
- Cobirka, M.; Tancin, V.; Slama, P. Epidemiology and Classification of Mastitis. Animals 2020, 10, 2212. [Google Scholar] [CrossRef] [PubMed]
- Bolte, J.; Zhang, Y.; Wente, N.; Krömker, V. In Vitro Susceptibility of Mastitis Pathogens Isolated from Clinical Mastitis Cases on Northern German Dairy Farms. Vet. Sci. 2020, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Dalanezi, F.M.; Joaquim, S.F.; Guimarães, F.F.; Guerra, S.T.; Lopes, B.C.; Schmidt, E.M.S.; Cerri, R.L.A.; Langoni, H. Influence of pathogens causing clinical mastitis on reproductive variables of dairy cows. J. Dairy Sci. 2020, 103, 3648–3655. [Google Scholar] [CrossRef] [PubMed]
- Traversari, J.; van den Borne, B.H.P.; Dolder, C.; Thomann, A.; Perreten, V.; Bodmer, M. Non-aureus Staphylococci Species in the Teat Canal and Milk in Four Commercial Swiss Dairy Herds. Front. Vet. Sci. 2019, 6, 186. [Google Scholar] [CrossRef] [Green Version]
- Siebert, A.; Hofmann, K.; Staib, L.; Doll, E.V.; Scherer, S.; Wenning, M. Amplicon-sequencing of raw milk microbiota: Impact of DNA extraction and library-PCR. Appl. Microbiol. Biotechnol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Godon, J.J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. Msphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Cendron, F.; Niero, G.; Carlino, G.; Penasa, M.; Cassandro, M. Characterizing the fecal bacteria and archaea community of heifers and lactating cows through 16S rRNA next-generation sequencing. J. Appl. Genet. 2020, 61, 593–605. [Google Scholar] [CrossRef]
- Zhu, Z.; Kristensen, L.; Difford, G.F.; Poulsen, M.; Noel, S.J.; Abu Al-Soud, W.; Sørensen, S.J.; Lassen, J.; Løvendahl, P.; Højberg, O. Changes in rumen bacterial and archaeal communities over the transition period in primiparous Holstein dairy cows. J. Dairy Sci. 2018, 101, 9847–9862. [Google Scholar] [CrossRef] [PubMed]
- Dworecka-Kaszak, B.; Krutkiewicz, A.; Szopa, D.; Kleczkowski, M.; Biegańska, M. High prevalence of Candida yeast in milk samples from cows suffering from mastitis in poland. Sci. World J. 2012, 2012, 196347. [Google Scholar] [CrossRef] [Green Version]
- Spanamberg, A.; Augusto Wünder, E.; Isabel Brayer Pereira, D.; Argenta, J.; Maria Cavallini Sanches, E.; Valente, P.; Ferreiro, L. Etiología de la mastitis bovina producida por levaduras en el sur de Brasil. Rev. Iberoam. De Micol. 2008, 25, 154–156. [Google Scholar] [CrossRef]
- Usman, T.; Yu, Y.; Liu, C.; Fan, Z.; Wang, Y. Comparison of methods for high quantity and quality genomic DNA extraction from raw cow milk. Genet. Mol. Res. 2014, 13, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Kennang Ouamba, A.J.; LaPointe, G.; Dufour, S.; Roy, D. Optimization of Preservation Methods Allows Deeper Insights into Changes of Raw Milk Microbiota. Microorganisms 2020, 8, 368. [Google Scholar] [CrossRef] [Green Version]
- Dahlberg, J.; Sun, L.; Persson Waller, K.; Östensson, K.; McGuire, M.; Agenäs, S.; Dicksved, J. Microbiota data from low biomass milk samples is markedly affected by laboratory and reagent contamination. PLoS ONE 2019, 14, e0218257. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Kable, M.E.; Marco, M.L. Impact of DNA Sequencing and Analysis Methods on 16S rRNA Gene Bacterial Community Analysis of Dairy Products. Msphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.W.; Martin, J.C.; Scott, P.; Parkhill, J.; Flint, H.J.; Scott, K.P. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 2015, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- McHugh, A.J.; Yap, M.; Crispie, F.; Feehily, C.; Hill, C.; Cotter, P.D. Microbiome-based environmental monitoring of a dairy processing facility highlights the challenges associated with low microbial-load samples. NPJ Sci. Food 2021, 5, 4. [Google Scholar] [CrossRef]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.S.; Andersen, K.S.; McIlroy, S.J.; Kristensen, J.M.; Yashiro, E.; Karst, S.M.; Albertsen, M.; Nielsen, P.H. Generation of Comprehensive Ecosystem-Specific Reference Databases with Species-Level Resolution by High-Throughput Full-Length 16S rRNA Gene Sequencing and Automated Taxonomy Assignment (AutoTax). MBio 2020, 11, e01557-20. [Google Scholar] [CrossRef]
- Escapa, I.F.; Huang, Y.; Chen, T.; Lin, M.; Kokaras, A.; Dewhirst, F.E.; Lemon, K.P. Construction of habitat-specific training sets to achieve species-level assignment in 16S rRNA gene datasets. Microbiome 2020, 8, 65. [Google Scholar] [CrossRef]
- Jeong, J.; Yun, K.; Mun, S.; Chung, W.-H.; Choi, S.-Y.; Nam, Y.-d.; Lim, M.Y.; Hong, C.P.; Park, C.; Ahn, Y.; et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci. Rep. 2021, 11, 1727. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, G.; Bicalho, M.L.; Meira, E.; Rossi, R.E.; Foditsch, C.; Machado, V.S.; Teixeira, A.G.V.; Santisteban, C.; Schukken, Y.H.; Bicalho, R.C. Microbiota of Cow’s Milk; Distinguishing Healthy, Sub-Clinically and Clinically Diseased Quarters. PLoS ONE 2014, 9, e85904. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, J.S.; Gorden, P.J.; Munro, D.; Rong, R.; Dong, Q.; Plummer, P.J.; Wang, C.; Phillips, G.J. Bacterial community profiling of milk samples as a means to understand culture-negative bovine clinical mastitis. PLoS ONE 2013, 8, e61959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nan, X.; Zhao, Y.; Jiang, L.; Wang, M.; Wang, H.; Zhang, F.; Xue, F.; Hua, D.; Liu, J.; et al. Rumen microbiome structure and metabolites activity in dairy cows with clinical and subclinical mastitis. J. Anim. Sci. Biotechnol. 2021, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Hamel, J.; Zhang, Y.; Wente, N.; Krömker, V. Non-S. aureus staphylococci (NAS) in milk samples: Infection or contamination? Vet. Microbiol. 2020, 242, 108594. [Google Scholar] [CrossRef]
- Zaragoza, C.S.; Olivares, R.A.; Watty, A.E.; Moctezuma Ade, L.; Tanaca, L.V. Yeasts isolation from bovine mammary glands under different mastitis status in the Mexican High Plateu. Rev. Iberoam Micol. 2011, 28, 79–82. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, Y.; Liu, H.; Zhao, S.; Wang, J.; Zheng, N. Characterization of Pseudomonas spp. and Associated Proteolytic Properties in Raw Milk Stored at Low Temperatures. Front. Microbiol. 2017, 8, 2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, C.; Hofmann, K.; Huptas, C.; Scherer, S.; Wenning, M.; Lücking, G. Simultaneous quantification of the most common and proteolytic Pseudomonas species in raw milk by multiplex qPCR. Appl. Microbiol. Biotechnol. 2021, 105, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from -omics studies. Mol. Biosyst. 2016, 12, 2359–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Total Bacterial Count (CFU/mL) | Individual Bacterial Count (IBC/mL) | Somatic Cell Count (SCC) per mL (Average Values Detected for Last 12 Months) |
|---|---|---|---|
| 796 | 6.00 × 104 | 2.23 × 105 | not available |
| 797 | 1.23 × 105 | 4.69 × 105 | not available |
| 798 | 1.55 × 105 | 5.97 × 105 | not available |
| 879 | 5.30 × 104 | 1.95 × 105 | 247,000 |
| 880 | 2.70 × 104 | 9.80 × 104 | 149,000 |
| 978 | 2.90 × 104 | 1.02 × 105 | 146,000 |
| 979 | 2.30 × 104 | 8.20 × 104 | 90,000 |
| 980 | 2.00 × 104 | 7.10 × 104 | 185,000 |
| 981 | 3.20 × 104 | 1.15 × 105 | 91,000 |
| 983 | 9.00 × 103 | 3.20 × 104 | 96,000 |
| Top Hit | Archaea | Eukaryotes |
|---|---|---|
| Top 1 | Methanobrevibacter sp. | Pichia scutulata |
| Top 2 | Methanobrevibacter millerae | Saprochaete clavata |
| Top 3 | Methanosarcina soligelidi | Pichia cactophila |
| Top 4 | Methanosarcina mazei | Kluyveromyces marxianus |
| Top 5 | Methanosarcina horonobensis | Pichia pseudocactophila |
| Species | Found in Raw Milk Samples | Detected on Genus Level in Raw Milk Samples | Found in Mock Communities |
|---|---|---|---|
| Arcanobacterium/ Truperella pyogenes | yes | Trueperella | no |
| Corynebacterium bovis | no | Corynebacterium | no |
| Enterobacter aerogenes | no | no | no |
| Enterococcus durans | yes | Enterococcus | no |
| Enterococcus faecalis | yes | no | |
| Enterococcus faecium | yes | no | |
| Escherichia coli | yes | Escherichia/Shigella | Escherichia coli |
| Klebsiella oxytoca | yes | Klebsiella | no |
| Klebsiella pneumoniae | no | Klebsiella pneumoniae | |
| Mycoplasma bovis | no | no | no |
| Proteus spp. (*) | no | no | no |
| Pseudomonas aeruginosa | no | Pseudomonas | Pseudomonas aeruginosa |
| Serratia marcescens(*) | yes | Serratia | no |
| Staphylococcus aureus | yes | Staphylococcus | Staphylococcus aureus |
| Staphylococcus chromogenes | yes | no | |
| Staphylococcus epidermidis | yes | Staphylococcus epidermidis | |
| Staphylococcus haemolyticus | yes | no | |
| Staphylococcus sciuri | yes | no | |
| Staphylococcus simulans | yes | no | |
| Streptococcus agalactiae | yes | Streptococcus | no |
| Streptococcus bovis | no | no | |
| Streptococcus dysgalactiae | yes | no | |
| Streptococcus equinus | yes | no | |
| Streptococcus uberis | yes | no | |
| Yersinia spp. (*) | no | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abellan-Schneyder, I.; Siebert, A.; Hofmann, K.; Wenning, M.; Neuhaus, K. Full-Length SSU rRNA Gene Sequencing Allows Species-Level Detection of Bacteria, Archaea, and Yeasts Present in Milk. Microorganisms 2021, 9, 1251. https://doi.org/10.3390/microorganisms9061251
Abellan-Schneyder I, Siebert A, Hofmann K, Wenning M, Neuhaus K. Full-Length SSU rRNA Gene Sequencing Allows Species-Level Detection of Bacteria, Archaea, and Yeasts Present in Milk. Microorganisms. 2021; 9(6):1251. https://doi.org/10.3390/microorganisms9061251
Chicago/Turabian StyleAbellan-Schneyder, Isabel, Annemarie Siebert, Katharina Hofmann, Mareike Wenning, and Klaus Neuhaus. 2021. "Full-Length SSU rRNA Gene Sequencing Allows Species-Level Detection of Bacteria, Archaea, and Yeasts Present in Milk" Microorganisms 9, no. 6: 1251. https://doi.org/10.3390/microorganisms9061251
APA StyleAbellan-Schneyder, I., Siebert, A., Hofmann, K., Wenning, M., & Neuhaus, K. (2021). Full-Length SSU rRNA Gene Sequencing Allows Species-Level Detection of Bacteria, Archaea, and Yeasts Present in Milk. Microorganisms, 9(6), 1251. https://doi.org/10.3390/microorganisms9061251