
Predicting Neuroblastoma Patient Risk Groups, Outcomes, and Treatment Response Using Machine Learning Methods: A Review

Abstract
:1. Introduction
2. The Use of Patient Data for Predicting Patient Outcomes
2.1. The Use of Expression-Based Data for the Development of Multi-Gene Predictor Models and INSS Staging
2.2. Methylome Data to Predict MYCN Status-Linked Outcomes
2.3. Histological Data for ML to Assist NB Diagnostics
2.4. Radiological Data for Clinical Predictions
3. ML for 3 Critical Clinical Aspects (Risk, Outcomes including Survival, and Treatment)
3.1. ML to Determine Risk Stratification
3.2. ML to Predict NB Patient Outcomes, including Survival
3.3. ML to Predict NB Response to Treatment
4. Discussion
4.1. Multi-Omics Data and Relevant Tools for Predicting NB Clinical Aspects
4.2. Histological Data and Relevant Tools for Predicting Clinical Aspects
4.3. Medical Imaging Data and Relevant Tools for Predicting NB Clinical Aspects
4.4. Investigating Clinicopathological Aspects of NB Patients
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALT | Alternating length of telomerase |
| ANN | Artificial neural network |
| CAD | Computer-aided diagnostics |
| CIMP | CpG island methylator phenotype |
| CNN | Convolutional neural network |
| DL | Deep learning |
| DNN | Deep neural network |
| EFS | Event-free survival |
| GEO | Gene expression omnibus |
| HFS | Heterogenous feature selection |
| HR | Hazard ratio |
| IDRF | Image-defined risk factors |
| INPC | International neuroblastoma pathology classification |
| INSS | International neuroblastoma staging system |
| INRGSS | International neuroblastoma risk group staging system |
| MIBG | Metaiodobenzylguanidine |
| MKI | Mitosis-karyorrhexis index |
| ML | Machine learning |
| MLP | Multi-layer perception |
| OS | Overall survival |
| PFS | Progression-free survival |
| RFE | Recursive feature elimination |
| SIFT | Scale Invariant Feature Transform |
| TARGET | Therapeutically applicable research to generate effective treatment |
| TCGA | The cancer genome atlas databases |
| TDA | Topological data analysis |
| TERT | Telomerase reverse transcriptase |
| TMA | Tissue microarrays |
| OVR | One-versus-rest |
| UMAP | Uniform manifold approximation and projection |
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Maris, J.M.; Yamashiro, D.J.; Hogarty, M.D.; White, P.S. Biology and genetics of human neuroblastomas. J. Pediatr. Hematol. Oncol. 1997, 19, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Cecchetto, G.; Mosseri, V.; De Bernardi, B.; Helardot, P.; Monclair, T.; Costa, E.; Horcher, E.; Neuenschwander, S.; Tomà, P.; Rizzo, A.; et al. Surgical risk factors in primary surgery for localized neuroblastoma: The LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J. Clin. Oncol. 2005, 23, 8483–8489. [Google Scholar] [CrossRef]
- Castleberry, R.P.; Pritchard, J.; Ambros, P.; Berthold, F.; Brodeur, G.M.; Castel, V.; Cohn, S.L.; De Bernardi, B.; Dicks-Mireaux, C.; Frappaz, D.; et al. The International Neuroblastoma Risk Groups (INRG): A preliminary report. Eur. J. Cancer 1997, 33, 2113–2116. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Perez, J.; Poon, C.C.Y.; Merrifield, R.D.; Wong, S.T.C.; Yang, G.-Z. Big data for health. IEEE J. Biomed. Health Inform. 2015, 19, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zeng, L.; Jin, X.; Lin, H.; Song, J. Feature Genes in Neuroblastoma Distinguishing High-Risk and Non-high-Risk Neuroblastoma Patients: Development and Validation Combining Random Forest With Artificial Neural Network. Front. Med. 2022, 9, 882348. [Google Scholar] [CrossRef] [PubMed]
- Deaton, C. Outcomes measurement. J. Cardiovasc. Nurs. 1998, 12, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, J.; De Preter, K.; Naranjo, A.; Vercruysse, L.; Van Roy, N.; Hellemans, J.; Swerts, K.; Bravo, S.; Scaruffi, P.; Tonini, G.P.; et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: A retrospective SIOPEN/COG/GPOH study. Lancet. Oncol. 2009, 10, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Wei, J.S.; Ringnér, M.; Saal, L.H.; Ladanyi, M.; Westermann, F.; Berthold, F.; Schwab, M.; Antonescu, C.R.; Peterson, C.; et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001, 7, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Sertel, O.; Shimada, H.; Boyer, K.L.; Saltz, J.H.; Gurcan, M.N. Computer-aided evaluation of neuroblastoma on whole-slide histology images: Classifying grade of neuroblastic differentiation. Pattern Recognit. 2009, 42, 1080–1092. [Google Scholar] [CrossRef]
- Liu, G.; Poon, M.; Zapala, M.A.; Temple, W.C.; Vo, K.T.; Matthay, K.K.; Mitra, D.; Seo, Y. Incorporating Radiomics into Machine Learning Models to Predict Outcomes of Neuroblastoma. J. Digit. Imaging 2022, 35, 605–612. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, B.; Yoon, S.W.; Ko, H.S. A support vector machine-based ensemble algorithm for breast cancer diagnosis. Eur. J. Oper. Res. 2018, 267, 687–699. [Google Scholar] [CrossRef]
- Zhao, L.; Feng, D. Deep Neural Networks for Survival Analysis Using Pseudo Values. IEEE J. Biomed. Health Inform. 2020, 24, 3308–3314. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef]
- Alam, M.Z.; Rahman, M.S.; Rahman, M.S. A Random Forest based predictor for medical data classification using feature ranking. Inform. Med. Unlocked 2019, 15, 100180. [Google Scholar] [CrossRef]
- Oberthuer, A.; Juraeva, D.; Hero, B.; Volland, R.; Sterz, C.; Schmidt, R.; Faldum, A.; Kahlert, Y.; Engesser, A.; Asgharzadeh, S.; et al. Revised risk estimation and treatment stratification of low- and intermediate-risk neuroblastoma patients by integrating clinical and molecular prognostic markers. Clin. Cancer Res. 2015, 21, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, H.; Darestani, S.A.; Azimi, P. An artificial neural network based mathematical model for a stochastic health care facility location problem. Health Care Manag. Sci. 2021, 24, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Delen, D.; Walker, G.; Kadam, A. Predicting breast cancer survivability: A comparison of three data mining methods. Artif. Intell. Med. 2005, 34, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Rigatti, S.J. Random Forest. J. Insur. Med. 2017, 47, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Mayampurath, A.; Ramesh, S.; Michael, D.; Liu, L.; Feinberg, N.; Granger, M.; Naranjo, A.; Cohn, S.L.; Volchenboum, S.L.; Applebaum, M.A. Predicting Response to Chemotherapy in Patients With Newly Diagnosed High-Risk Neuroblastoma: A Report From the International Neuroblastoma Risk Group. JCO Clin. Cancer Inform. 2021, 5, 1181–1188. [Google Scholar] [CrossRef]
- Park, A.; Nam, S. Deep learning for stage prediction in neuroblastoma using gene expression data. Genom. Inf. 2019, 17, e30. [Google Scholar] [CrossRef]
- Bi, W.L.; Hosny, A.; Schabath, M.B.; Giger, M.L.; Birkbak, N.J.; Mehrtash, A.; Allison, T.; Arnaout, O.; Abbosh, C.; Dunn, I.F.; et al. Artificial intelligence in cancer imaging: Clinical challenges and applications. CA Cancer J. Clin. 2019, 69, 127–157. [Google Scholar] [CrossRef]
- Kogner, P.; Barbany, G.; Björk, O.; Castello, M.A.; Donfrancesco, A.; Falkmer, U.G.; Hedborg, F.; Kouvidou, H.; Persson, H.; Raschella, G. Trk mRNA and low affinity nerve growth factor receptor mRNA expression and triploid DNA content in favorable neuroblastoma tumors. Prog. Clin. Biol. Res. 1994, 385, 137–145. [Google Scholar]
- Carén, H.; Erichsen, J.; Olsson, L.; Enerbäck, C.; Sjöberg, R.-M.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: Four cases of homozygous deletions of the CDKN2A gene. BMC Genom. 2008, 9, 353. [Google Scholar] [CrossRef]
- Abel, F.; Dalevi, D.; Nethander, M.; Jörnsten, R.; De Preter, K.; Vermeulen, J.; Stallings, R.; Kogner, P.; Maris, J.; Nilsson, S. A 6-gene signature identifies four molecular subgroups of neuroblastoma. Cancer Cell Int. 2011, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Mayol, G.; Ríos, J.; Domenech, G.; Cheung, N.-K.V.; Oberthuer, A.; Fischer, M.; Maris, J.M.; Brodeur, G.M.; Hero, B.; et al. A three-gene expression signature model for risk stratification of patients with neuroblastoma. Clin. Cancer Res. 2012, 18, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Durinck, K.; Speleman, F. Epigenetic regulation of neuroblastoma development. Cell Tissue Res. 2018, 372, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Decock, A.; Ongenaert, M.; Vandesompele, J.; Speleman, F. Neuroblastoma epigenetics: From candidate gene approaches to genome-wide screenings. Epigenetics 2011, 6, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Abe, M.; Ushijima, T. Clinical application of the CpG island methylator phenotype to prognostic diagnosis in neuroblastomas. J. Hum. Genet. 2013, 58, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Giwa, A.; Rossouw, S.C.; Fatai, A.; Gamieldien, J.; Christoffels, A.; Bendou, H. Predicting amplification of MYCN using CpG methylation biomarkers in neuroblastoma. Futur. Oncol. 2021, 17, 4769–4783. [Google Scholar] [CrossRef]
- Sugino, R.P.; Ohira, M.; Mansai, S.P.; Kamijo, T. Comparative epigenomics by machine learning approach for neuroblastoma. BMC Genom. 2022, 23, 852. [Google Scholar] [CrossRef]
- Gheisari, S.; Catchpoole, D.R.; Charlton, A.; Melegh, Z.; Gradhand, E.; Kennedy, P.J. Computer Aided Classification of Neuroblastoma Histological Images Using Scale Invariant Feature Transform with Feature Encoding. Diagnostics 2018, 8, 56. [Google Scholar] [CrossRef]
- Yu, K.-H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Y.; Hou, C.; Li, N.; Zhang, N.; Yan, X.; Yang, L.; Guo, Y.; Chen, H.; Li, J.; et al. Pathological prognosis classification of patients with neuroblastoma using computational pathology analysis. Comput. Biol. Med. 2022, 149, 105980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lv, C.; Jin, Y.; Cheng, G.; Fu, Y.; Yuan, D.; Tao, Y.; Guo, Y.; Ni, X.; Shi, T. Deep Learning-Based Multi-Omics Data Integration Reveals Two Prognostic Subtypes in High-Risk Neuroblastoma. Front. Genet. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Huang, R.; Stucky, A.; Chen, X.; Sun, L.; Wen, Q.; Zeng, Y.; Fletcher, H.; Wang, C.; et al. The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival. Cancers 2022, 14, 2874. [Google Scholar] [CrossRef] [PubMed]
- Cangelosi, D.; Pelassa, S.; Morini, M.; Conte, M.; Bosco, M.C.; Eva, A.; Sementa, A.R.; Varesio, L. Artificial neural network classifier predicts neuroblastoma patients’ outcome. BMC Bioinform. 2016, 17, 347. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, L.; Ishola, T.; Pucci, P.; Trigg, R.M.; Pereira, J.; Williams, J.A.; Cavanagh, M.L.; Gkoutos, G.V.; Tsaprouni, L.; Turner, S.D. The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells. Cancers 2021, 13, 1239. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, L. Profiling of the Prognostic Role of Extracellular Matrix-Related Genes in Neuroblastoma Using Databases and Integrated Bioinformatics. Onco 2022, 2, 85–112. [Google Scholar] [CrossRef]
- Popov, A.M.; Shorikov, E.V.; Verzhditskaia, T.I.; Tsaur, G.A.; Druĭ, A.E.; Solodovnikov, A.G.; Savel’ev, L.I.; Fechina, L.G. Prognostic value of bone marrow lesions in children with neuroblastoma detected by flow cytometry. Vopr. Onkol. 2014, 60, 469–475. [Google Scholar] [PubMed]
- Wei, J.S.; Greer, B.T.; Westermann, F.; Steinberg, S.M.; Son, C.-G.; Chen, Q.-R.; Whiteford, C.C.; Bilke, S.; Krasnoselsky, A.L.; Cenacchi, N.; et al. Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res. 2004, 64, 6883–6891. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, X.; Zhang, J. A Heterogeneous Ensemble Learning Method For Neuroblastoma Survival Prediction. IEEE J. Biomed. Health Inform. 2022, 26, 1472–1483. [Google Scholar] [CrossRef]
- Ghaderi Zefrehi, H.; Altınçay, H. Imbalance learning using heterogeneous ensembles. Expert Syst. Appl. 2020, 142, 113005. [Google Scholar] [CrossRef]
- Feng, C.; Xiang, T.; Yi, Z.; Meng, X.; Chu, X.; Huang, G.; Zhao, X.; Chen, F.; Xiong, B.; Feng, J. A Deep-Learning Model With the Attention Mechanism Could Rigorously Predict Survivals in Neuroblastoma. Front. Oncol. 2021, 11, 653863. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Gong, J.; Lin, P.; Zhang, M.; Wu, K. Machine learning methods revealed the roles of immune-metabolism related genes in immune infiltration, stemness, and prognosis of neuroblastoma. Cancer Biomark. 2023, 38, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Björklund, M. Be careful with your principal components. Evolution 2019, 73, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet. Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lue, W.; Kaalia, R.; Kumar, P.; Rajapakse, J.C. Network-based integration of multi-omics data for clinical outcome prediction in neuroblastoma. Sci. Rep. 2022, 12, 15425. [Google Scholar] [CrossRef] [PubMed]
- Gheisari, S.; Catchpoole, D.R.; Charlton, A.; Kennedy, P.J. Convolutional Deep Belief Network with Feature Encoding for Classification of Neuroblastoma Histological Images. J. Pathol. Inform. 2018, 9, 17. [Google Scholar] [CrossRef]
- Bussola, N.; Papa, B.; Melaiu, O.; Castellano, A.; Fruci, D.; Jurman, G. Quantification of the Immune Content in Neuroblastoma: Deep Learning and Topological Data Analysis in Digital Pathology. Int. J. Mol. Sci. 2021, 22, 8804. [Google Scholar] [CrossRef]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Huang, K.; Liu, H.; Ding, H.; Zhang, L.; Zhang, T.; Yu, W.; He, L. CT-Based Radiomics Signature With Machine Learning Predicts MYCN Amplification in Pediatric Abdominal Neuroblastoma. Front. Oncol. 2021, 11, 687884. [Google Scholar] [CrossRef]
- Florkow, M.C.; Guerreiro, F.; Zijlstra, F.; Seravalli, E.; Janssens, G.O.; Maduro, J.H.; Knopf, A.C.; Castelein, R.M.; van Stralen, M.; Raaymakers, B.W.; et al. Deep learning-enabled MRI-only photon and proton therapy treatment planning for paediatric abdominal tumours. Radiother. Oncol. 2020, 153, 220–227. [Google Scholar] [CrossRef]
- Zormpas-Petridis, K.; Poon, E.; Clarke, M.; Jerome, N.P.; Boult, J.K.R.; Blackledge, M.D.; Carceller, F.; Koers, A.; Barone, G.; Pearson, A.D.J.; et al. Noninvasive MRI Native T(1) Mapping Detects Response to MYCN-targeted Therapies in the Th-MYCN Model of Neuroblastoma. Cancer Res. 2020, 80, 3424–3435. [Google Scholar] [CrossRef]
- Giwa, A.; Fatai, A.; Gamieldien, J.; Christoffels, A.; Bendou, H. Identification of novel prognostic markers of survival time in high-risk neuroblastoma using gene expression profiles. Oncotarget 2020, 11, 4293–4305. [Google Scholar] [CrossRef]
- Belthangady, C.; Royer, L.A. Applications, promises, and pitfalls of deep learning for fluorescence image reconstruction. Nat. Methods 2019, 16, 1215–1225. [Google Scholar] [CrossRef]
- Tranchevent, L.-C.; Azuaje, F.; Rajapakse, J.C. A deep neural network approach to predicting clinical outcomes of neuroblastoma patients. BMC Med. Genom. 2019, 12, 178. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 Expression in Neuroblastoma via Computational Modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef]
- Pirone, D.; Montella, A.; Sirico, D.G.; Mugnano, M.; Villone, M.M.; Bianco, V.; Miccio, L.; Porcelli, A.M.; Kurelac, I.; Capasso, M.; et al. Label-free liquid biopsy through the identification of tumor cells by machine learning-powered tomographic phase imaging flow cytometry. Sci. Rep. 2023, 13, 6042. [Google Scholar] [CrossRef]
- Martí-Bonmatí, L.; Alberich-Bayarri, Á.; Ladenstein, R.; Blanquer, I.; Segrelles, J.D.; Cerdá-Alberich, L.; Gkontra, P.; Hero, B.; García-Aznar, J.M.; Keim, D.; et al. PRIMAGE project: Predictive in silico multiscale analytics to support childhood cancer personalised evaluation empowered by imaging biomarkers. Eur. Radiol. Exp. 2020, 4, 22. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type Used and Lab Method | Tools Used | Findings | Reference |
|---|---|---|---|
| 59-gene signature obtained from data mining from 579 patient datasets (30 training, 313 testing, 236 validation), and qPCR | Data mining, multivariate Cox regression | Signature predictor of outcome; for example, patients with a higher risk signature, deemed at higher risk of death and relapse with an odds ratio for OS and PFS of 19.32 and 3.96. | [15] |
| 4 × 44 K microarray data from 709 NB specimens | SVM, Kaplan–Meier and Multivariate Cox regression | Classifiers showed the highest clinical value for low- and intermediate-risk patients (low-risk: EFS: 0.84, OS: 0.99, and intermediate-risk, EFS: 0.88, and OS: 1 for these groups. | [23] |
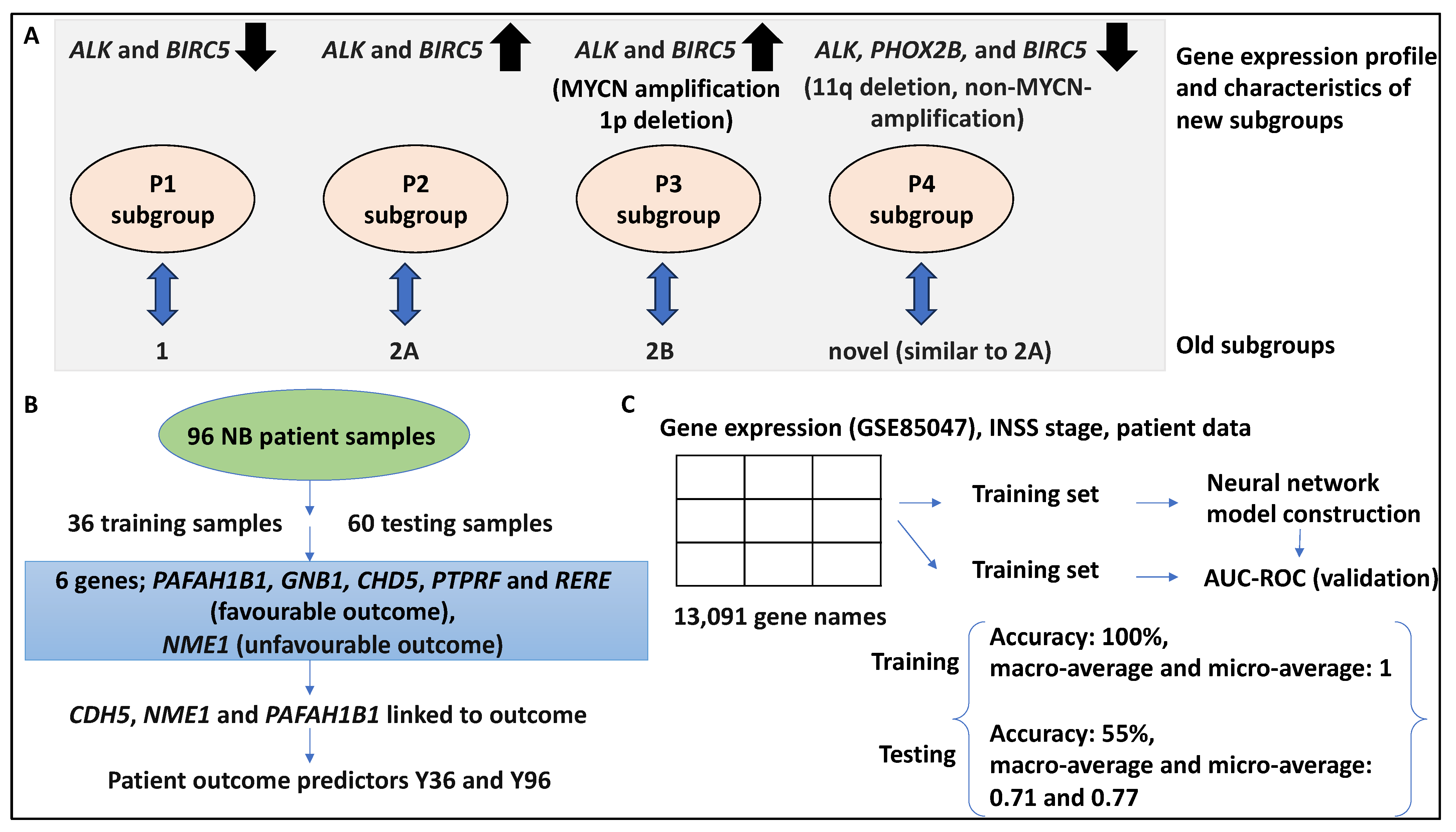
| 47 microarray samples (dataset 1 comprising 23 NB tumours, and dataset 2 comprising 30 NB tumours) and 101 NB samples for validation | Principal component analysis and unsupervised hierarchical clustering | 6-gene signature (ALK, BIRC5, MYCN, CCND1, NTRK1, and PHOX2B) identified 4 subgroups (p1–p4). Groups p1–p3 corresponded to subtypes 1,2A, and 2B, but p4 was novel (11q deletion, MYCN-non-amplified, low expression of ALK, BIRC5, and PHOX2B linked to poor outcomes). | [32] |
| 96 samples and tested on 362 separate microarray expression datasets, and RT-PCR | Univariate Cox regression and principal component analysis | A 3-gene (CDH5, PAFAH1B1, and NME1) expression signature for risk stratification, the Y36 predictor model could distinguish 2 groups in OS and EFS (HR, 9.3 and 3.1, respectively), Y96 was also formed. From the 352 validation samples, 2 groups with distinct OS and EFS were distinguished. | [33] |
| 280 NB datasets deposited in GSE85047 and its clinical data were obtained; matrices contained patient data (INSS stage and gene expression array) | DNN architecture | The OVR AUCs for patient stages ranging from 1–4S were 0.8, 0.66, 0.59, 0.85, and 0.58, respectively. | [28] |
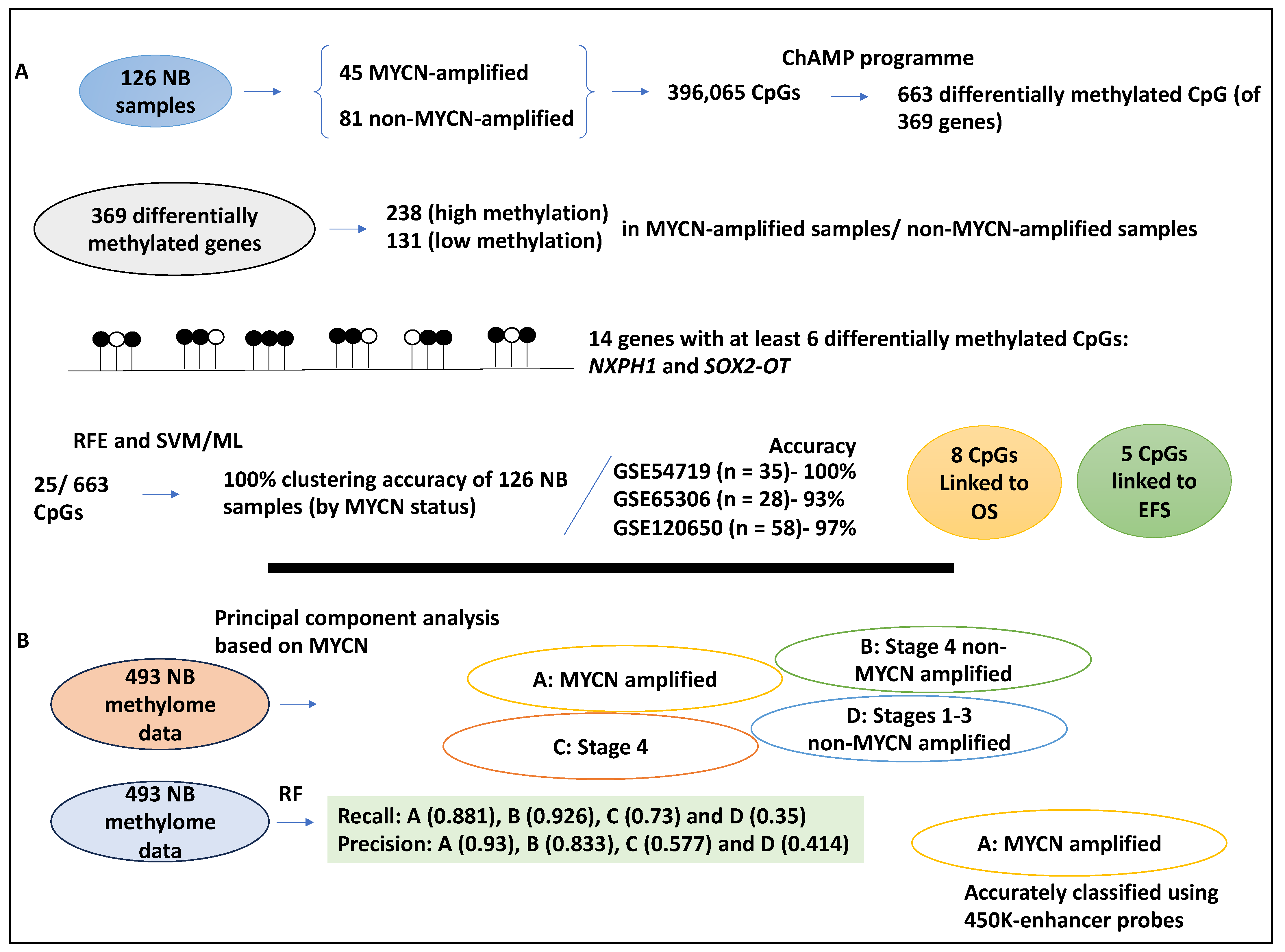
| 126 NB samples inclusive of 45 MYCN-amplified and 81 non-MYCN-amplified datasets and 663 differentially methylated CpGs were obtained | ChAMP programme, RFE, and ML (SVM), hierarchical and K-means clustering, Kaplan–Meier and Cox regression | 14 genes, including NXPH1 and SOX2-OT were highly methylated. 25/663 of these islands and the 663 CpGs led to correct clustering based on MYCN status. MYCN amplification status was associated with CpG score and patient survival (OS: HR = 5.11, EFS: HR = 4.84). | [37] |
| 493 NB methylome data referred to as the Human Methylation 450 K dataset | Principal component analysis and RF | Clustering based on MYCN led to 4 clusters: A, MYCN-amplified patients; B, stage 4 INSS without MYCN amplification; C, stage INSS 4 patients; and D, stage I-III without MYCN amplification. RF made accurate classifications for groups A and B (these groups were linked to DNA methylome). MYCN-amplified A group was linked to DNA methylation of the enhancer regions. | [38] |
| Training dataset consisted of 387 cropped image tiles obtained from 3 whole slides | Feature construction, selection, and extraction, classification by SVM, and leave-one-out method | Feature extraction followed by classification by SVM for resolution levels 1, 2, 3 and 4, yielded 3, 6, 10, and 5 features, respectively. Neuroblastic types comprising 10 undifferentiated, 13 differentiated, and 10 poorly differentiated NB, yielded accuracies of 90, 84.62 and 90%, respectively. | [17] |
| 6 TMA slides and 7 whole sections for 125 NB patients (3 datasets in total, 623 for training, 211 for the second section of the validation process, and 209 for testing). 623 sub-images and 5 whole sections for validation | SIFT with the bag of features and the SVM classifier | SIFT with the bag of features and the SVM classifier outperformed other methods with precision, recall, and F-measure were 83.81, 86.61, and 85.19, respectively. The algorithm first assigned a label and then classified using a majority vote. A tissue section (4905) was ganglioneuroma, and 10/10 sub-images were assigned correctly. | [39] |
| 563 H&E whole-slides were obtained from 107 NB patients with two distinct groups of favourable (67) and unfavourable (40) prognoses | Segmentation, feature extraction and per-patient feature aggregation for feature extraction. Feature reduction, feature selection and model construction for ML method | After nuclear instance segmentation, the number of nuclei, those nuclei identified by the algorithm, and false positives were established as 3408, 3407, and 46, respectively, and, a recall and precision of 98.62% and 98.65%, respectively. In both the training and testing datasets, clinicopathological factors such as nucleus morphology intensity of features, and age could accurately classify the patients with an AUC of 0.946. | [41] |
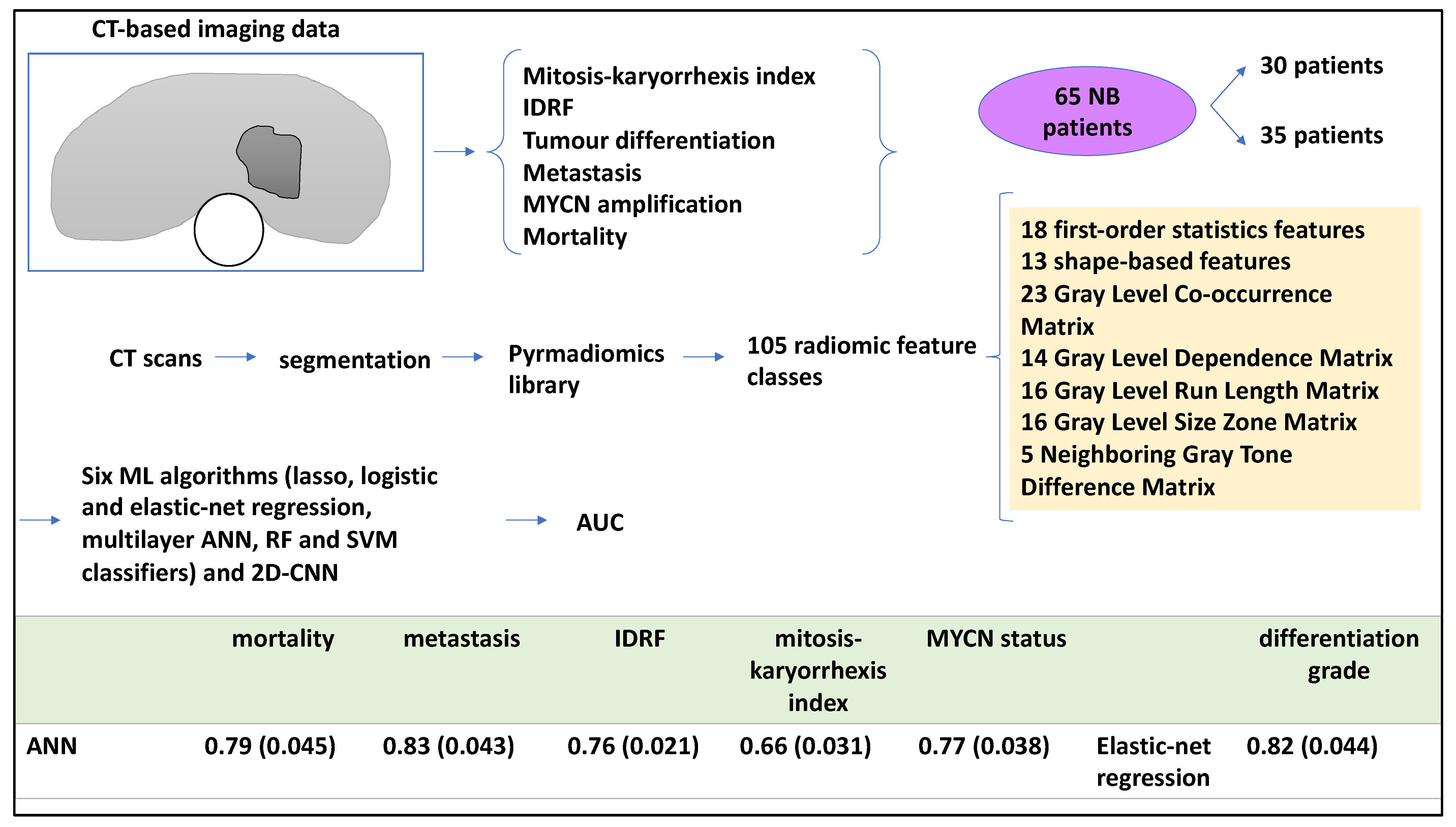
| 3D CT scans of 65 NB patients, primary tumours were segmented from CT scans and reviewed by a radiologist. A pyradiomics library was utilised to retrieve 105 radiomic features | Lasso, logistic and elastic-net regression, ANN, RF and SVM classifiers and 2D CNN | ANN obtained AUC-ROC of 0.79 (0.045) for mortality, 0.83 (0.043) for metastasis. 0.76 (0.021) for IDRF, 0.66 (0.031) for MKI index, and 0.77 (0.038) for MYCN status. For neuroblastic differentiation grade, elastic-net regression obtained and AUC of 0.82 (0.044). | [18] |
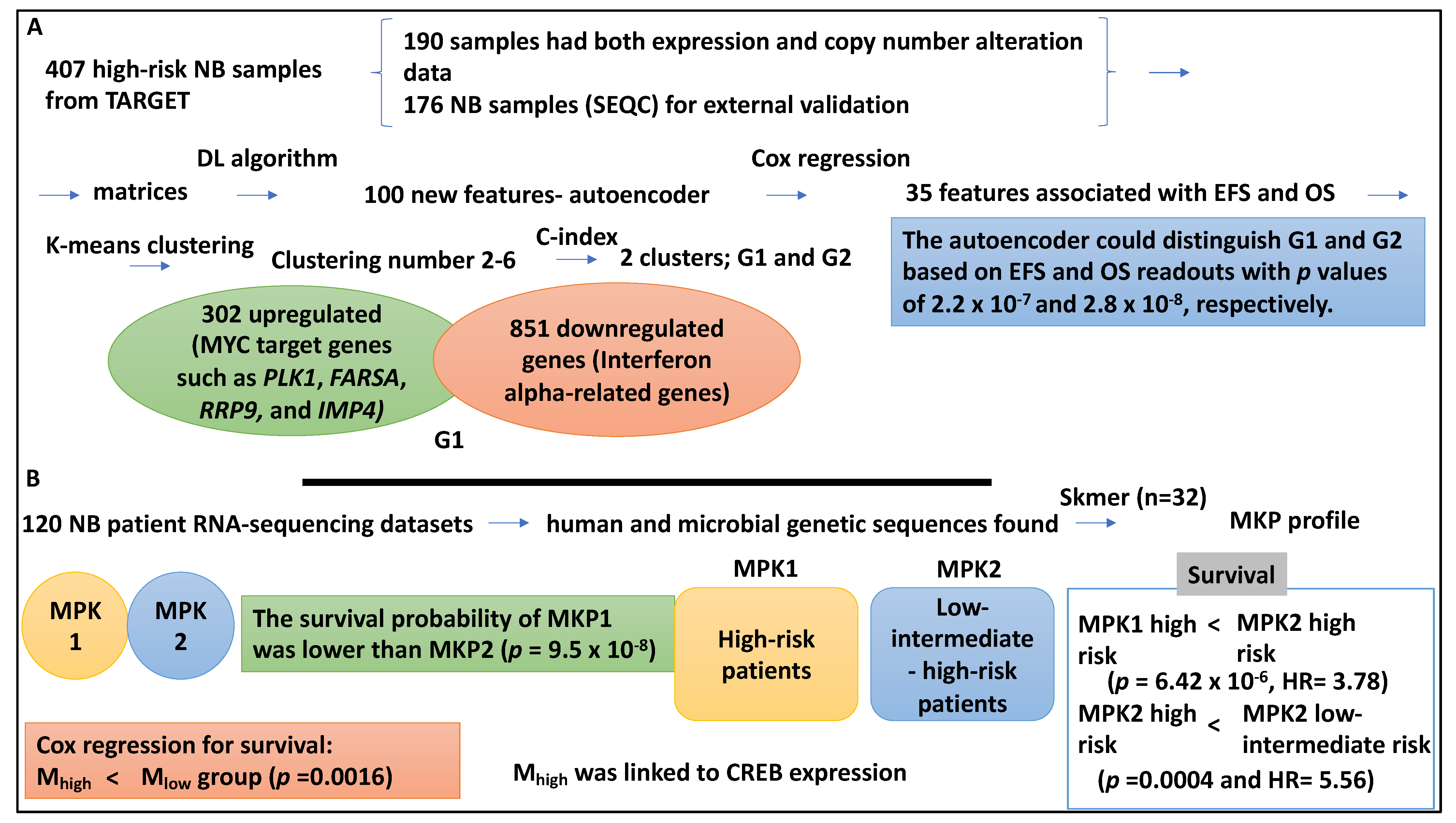
| 407 high-risk NB samples were collected from TARGET comprising 217 and 380 gene expression and copy number alteration datasets, 176 NB datasets for validation | DL (DNN), Cox regression, K-means clustering analysis, SVM, naïve Bayes, logistic regression and XGBoost | The autoencoder could distinguish G1 and G2 based on EFS and OS readouts with p values of 2.2 × 10−7 and 2.8 × 10−8, respectively. SVM performed better than the other three classifiers (average AUC of 0.844) and was also able to split the high-risk cases into two subgroups. MYC target genes such as PLK1, FARSA, RRP9, and IMP4 were upregulated in the G1 subtype (p value of 9.81 × 10−7). | [42] |
| 120 NB patient RNA-sequencing datasets, the mean age at diagnosis was four years and 3 months, and the majority were male and were classified as high-risk | Skmer, Pearson Chi-square test, Cox regression | Microbiome sequences; MKP1/2 profiles. The survival probability of MKP1 was lower than that of MKP2 (p = 9.5 × 10−8). The high-risk cases in the MKP1 group showed lower survival than their counterparts in MKP2 group (p = 6.422 × 10−6, HR = 3.78). High-risk NB patients in MKP2 also had lower survival than the low-intermediate-risk NB patients in MKP2 (p = 0.0004 and HR = 5.56). | [43] |
| 182 patient microarray datasets obtained from 4 cohorts (100 training and 82 testing) | ANN and leave-one-out method, Kaplan–Meier plots and log-rank tests | NB-hypo classifiers split the patients based on good and poor prognosis with distinct OS and EFS values (p-Value < 0.0001). Errors occurred when classifying stages 1–4 (including 4S), while 100% accuracy was obtained when processing low-intermediate-risk patients. | [44] |
| 56 treatment-naïve primary NB tumours (from 49 patients), 30 alive and 19 (dead), 37,920 clones selected | Principal component analysis, ANN, leave-one-out method, Kaplan–Meier analysis | The ANN could partition the high-risk patients based on 37,920 clones (p value of 0.0067) and 19 ANN-ranked genes (p value of 0.0005). | [48] |
| 1119 NB patient records with 31 variables, after preprocessing 1115 samples and 22 variables were selected. 689 patients were alive and the remaining were dead | DRGXG: heterogeneous ensemble learning strategy: HFS, heterogeneous base learners (DT, RF, SVM, and lightGBM, and XGBoost) and WAUCE | The earlier the diagnosis, the greater the likelihood that the prediction model would predict a “survival” outcome. The older the patient is at diagnosis, the greater the risk of “death” outcome. The larger the ploidy value, the greater the likelihood that the prediction model would assign “survival” as the status of the patient. | [49] |
| 721 NB patient microarray data, 172 features selected | Chi-square test, K-means clustering, supervised classification model (DNN) with attention mechanism | 50 genes marked the S1 group, and 122 genes marked the S2 group. MYCN amplification was associated with the S2 group. Multivariate Cox regression showed significance for MYCN status, age, risk, and stage (p > 0.05). Patients with MYCN amplification status, of any age, or gender, at stage 4 with high-risk classification were placed in the S2 group. | [51] |
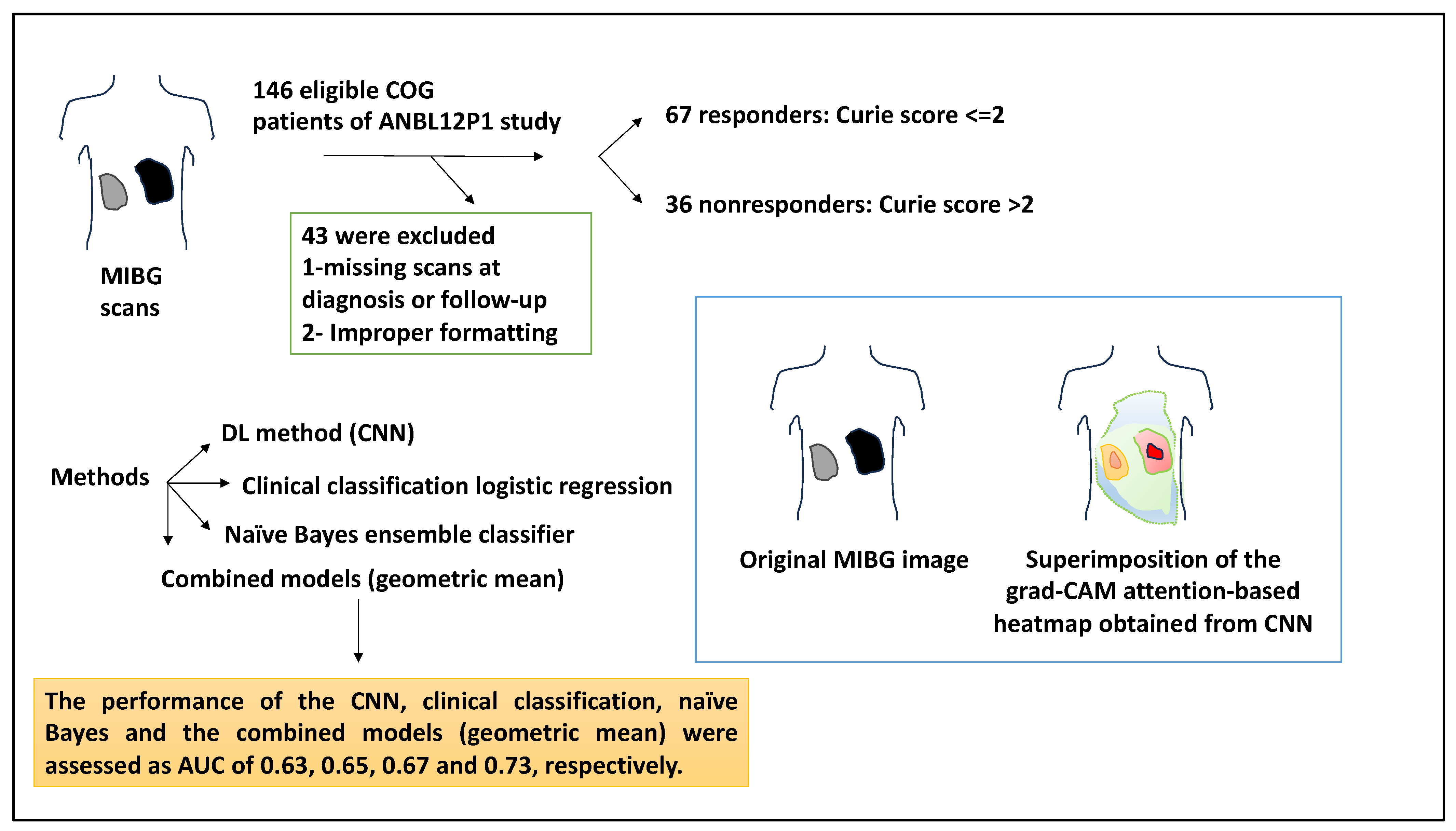
| 103 patients: patients with a Curie score ≤ 2 responded to induction chemotherapy (n = 67), whereas the nonresponders (n = 36) displayed a Curie score > 2 | DL (DNN) method (CNN), clinical classification logistic regression, naïve Bayes ensemble learner/classifier, and geometric models, Grad-CAM method | An original MIBG image and the same image superimposed with the grad-CAM attention-based heatmap obtained from the CNN analysis were shown. This composite utilised read colours for the areas that CNN paid the most attention to. The performance of the CNN, clinical classification, naïve Bayes, and the combined models (geometric mean) were AUC: 0.63, 0.65, 0.67, and 0.73, respectively. | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L. Predicting Neuroblastoma Patient Risk Groups, Outcomes, and Treatment Response Using Machine Learning Methods: A Review. Med. Sci. 2024, 12, 5. https://doi.org/10.3390/medsci12010005
Jahangiri L. Predicting Neuroblastoma Patient Risk Groups, Outcomes, and Treatment Response Using Machine Learning Methods: A Review. Medical Sciences. 2024; 12(1):5. https://doi.org/10.3390/medsci12010005
Chicago/Turabian StyleJahangiri, Leila. 2024. "Predicting Neuroblastoma Patient Risk Groups, Outcomes, and Treatment Response Using Machine Learning Methods: A Review" Medical Sciences 12, no. 1: 5. https://doi.org/10.3390/medsci12010005
APA StyleJahangiri, L. (2024). Predicting Neuroblastoma Patient Risk Groups, Outcomes, and Treatment Response Using Machine Learning Methods: A Review. Medical Sciences, 12(1), 5. https://doi.org/10.3390/medsci12010005