
Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer



Abstract
:1. Introduction
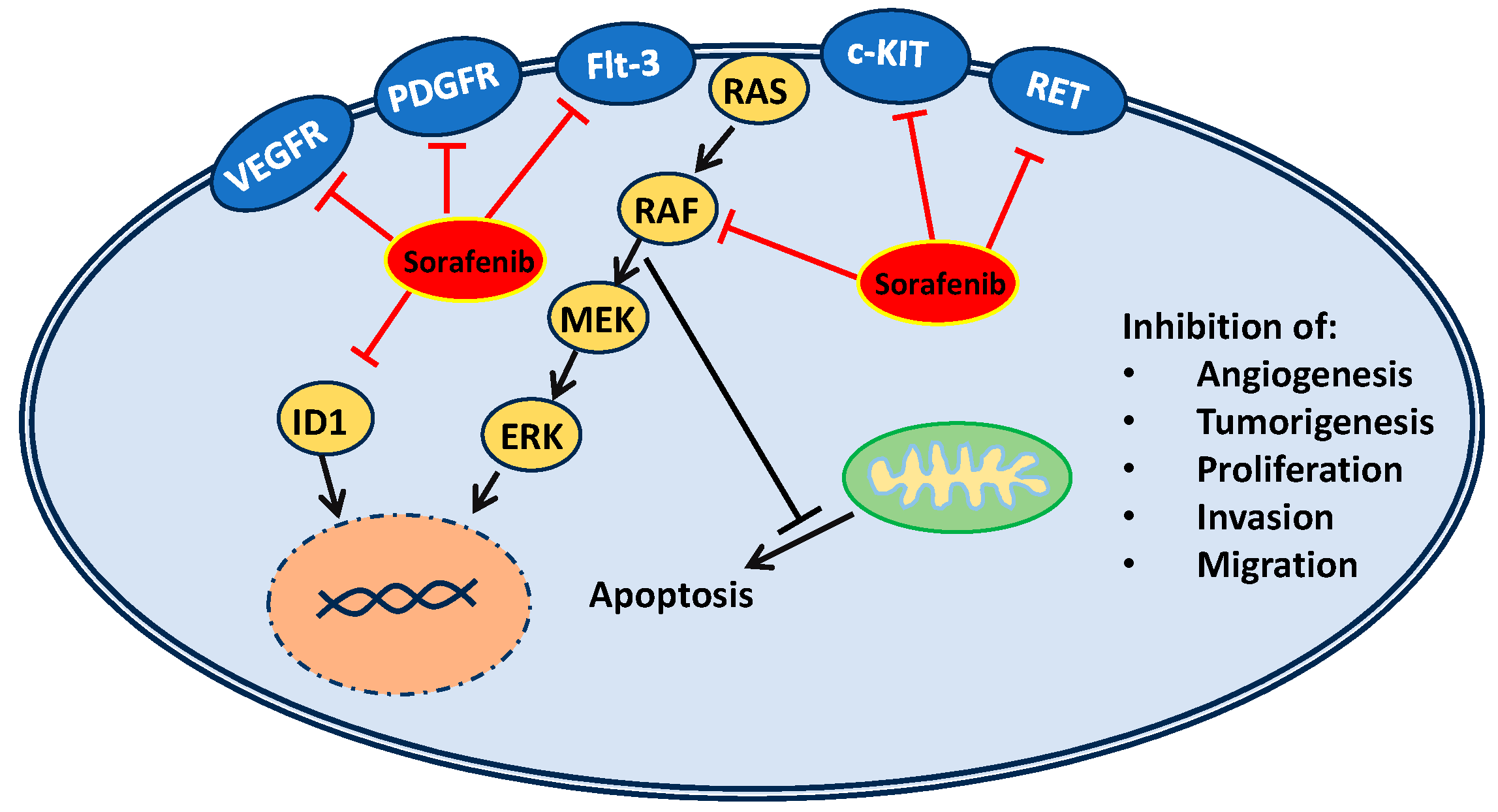
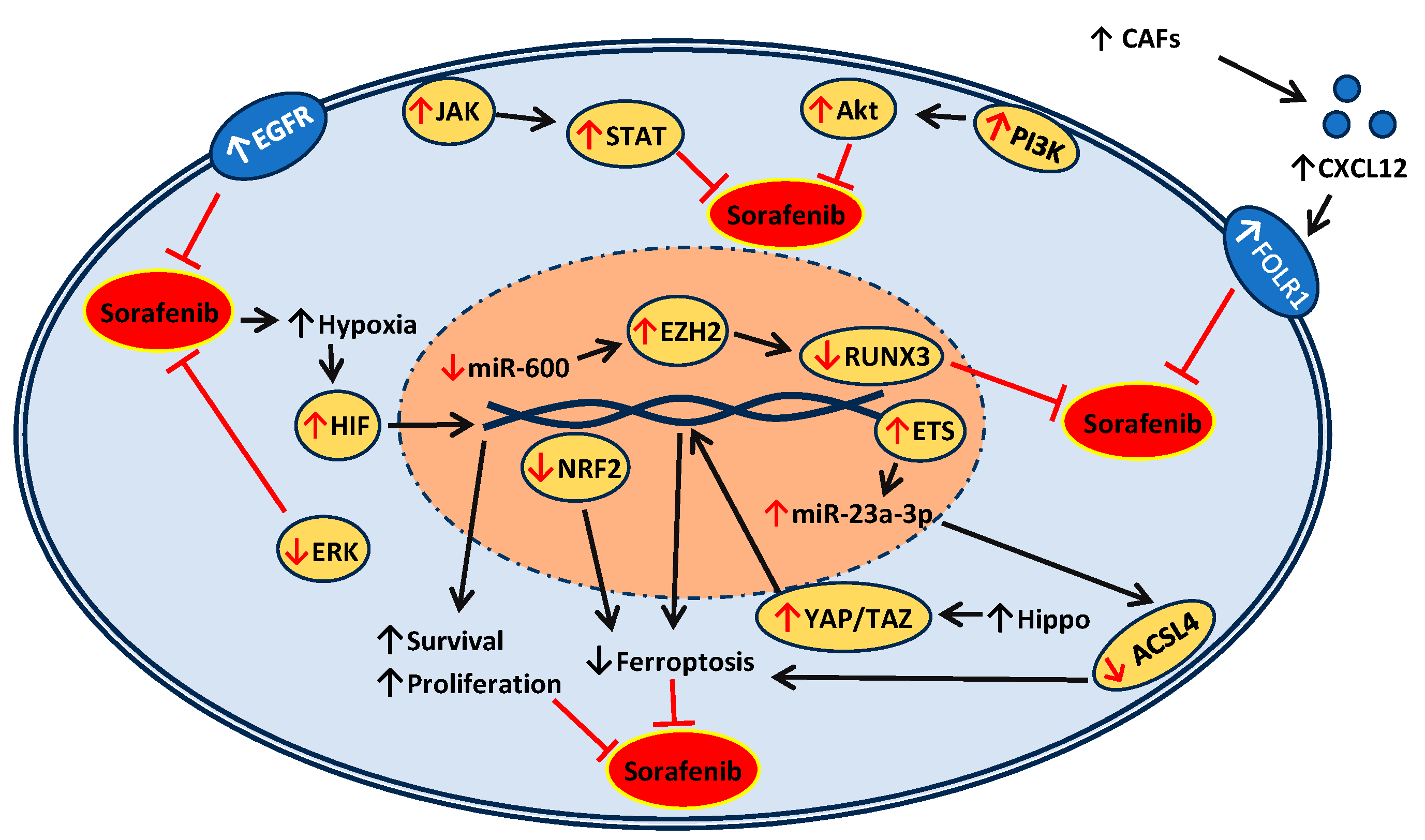
2. Mechanisms of Action and Resistance of Sorafenib
3. Studies of Sorafenib Efficacy in NSCLC
3.1. In Vitro and In Vivo Studies of Monotherapy and Dual-Therapy Approaches Involving Sorafenib
3.2. In Vitro and In Vivo Studies with Novel Approaches to Sorafenib Delivery
3.3. Clinical Studies Involving Sorafenib
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NSCLC | Non-small cell lung cancer |
| SCLC | Small cell lung cancer |
| VEGFR | Vascular endothelial growth factor receptor |
| PDGFR | Platelet-derived growth factor receptor |
| Flt-3 | FMS-like tyrosine kinase 3 |
| c-KIT | Type III receptor tyrosine kinase |
| RET | Rearranged during transfection |
| ID1 | Inhibitor of differentiation 1 |
| TKIs | Tyrosine kinase inhibitors |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| JAK | Janus Kinase |
| STAT | Signal transducers and activators of transcription |
| ETS1 | ETS Proto-Oncogene 1 |
| ACSL4 | acyl-CoA synthetase long chain family member 4 |
| CAFs | Cancer-associated fibroblasts |
| ccRCC | Clear cell renal cell carcinoma |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| RUNX3 | Runt-related transcription factor 3 |
| CAI | Carboxyamidotriazole |
| mTOR | Mammalian target of rapamycin |
| DCR | Disease control rates |
| OS | Overall survival |
| PFS | Progression-free survival |
| SSS | Sorafenib sensitivity signature |
| ORRs | Objective response rates |
| MTD | Maximum tolerated dose |
| PMR | Partial medical response |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Luo, L.; Wampfler, J.A.; Wang, Y.; Liu, D.; Chen, Y.-M.; Adjei, A.A.; Midthun, D.E.; Yang, P. 5-year overall survival in patients with lung cancer eligible or ineligible for screening according to US Preventive Services Task Force criteria: A prospective, observational cohort study. Lancet Oncol. 2019, 20, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Lung Cancer Fact Sheet|American Lung Association. Available online: https://www.lung.org/lung-health-diseases/lung-disease-lookup/lung-cancer/resource-library/lung-cancer-fact-sheet (accessed on 20 December 2023).
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.-P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.-Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE collaborative group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Herbst, R.S.; Wistuba, I.I.; Lee, J.J.; Blumenschein, G.R., Jr.; Tsao, A.; Stewart, D.J.; Hicks, M.E.; Erasmus, J., Jr.; Gupta, S.; et al. The BATTLE trial: Personalizing Therapy for Lung Cancer. Cancer Discov. 2011, 1, 44–53. [Google Scholar] [CrossRef]
- Pallis, A.; Serfass, L.; Dziadziuszko, R.; van Meerbeeck, J.; Fennell, D.; Lacombe, D.; Welch, J.; Gridelli, C. Targeted therapies in the treatment of advanced/metastatic NSCLC. Eur. J. Cancer 2009, 45, 2473–2487. [Google Scholar] [CrossRef]
- Liu SY, M.; Zheng, M.M.; Pan, Y.; Liu, S.Y.; Li, Y.; Wu, Y.L. Emerging evidence and treatment paradigm of non-small cell lung cancer. J. Hematol. Oncol. 2023, 16, 40. [Google Scholar] [CrossRef]
- Qu, J.; Wang, L.; Jiang, M.; Zhao, D.; Wang, Y.; Zhang, F.; Li, J.; Zhang, X. A review about pembrolizumab in first-line treatment of advanced NSCLC: Focus on KEYNOTE studies. Cancer Manag. Res. 2020, 12, 6493–6509. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for first-line treatment of PD-L1–selected patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. A. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Socinski, M.A.; Burns, T.F. Personalized treatment of EGFR mutant and ALK-positive patients in NSCLC. Expert Opin. Pharmacother. 2014, 15, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Rosell, R.; Morales-Espinosa, D.; Viteri, S. Systemic treatment in EGFR-ALK NSCLC patients: Second line therapy and beyond. Expert Rev. Anticancer Ther. 2014, 14, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Liu, J. Inhibitor of Differentiation 1 (ID1) Facilitates the Efficacy of Sorafenib in Non-Small Cell Lung Cancer Cells through Suppressing Epithelial to Mesenchymal Transition. Med. Sci. Monit. 2020, 26, e922148. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB summary: Sorafenib pathways. Pharmacogenet. Genom. 2017, 27, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Y.; Reiberger, T.; Duyverman, A.M.; Huang, P.; Samuel, R.; Hiddingh, L.; Roberge, S.; Koppel, C.; Lauwers, G.Y.; et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology 2014, 59, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl.) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Guo, L.; Hu, C.; Yao, M.; Han, G. Mechanism of sorafenib resistance associated with ferroptosis in HCC. Front. Pharmacol. 2023, 14, 1207496. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.S.; Zheng, Y.C.; Wang, N.; et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a- 3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, C.; Zhang, Y.; Tian, X.; Wang, H.; Wu, J.; Jiang, S. From synergy to resistance: Navigating the complex relationship between sorafenib and ferroptosis in hepatocellular carcinoma. Biomed. Pharmacother. 2024, 170, 116074. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, E.; Bai, Z.; Jia, Y.; Wang, B.; Dai, Y.; Zhuo, W.; Zeng, G.; Liu, X.; Cai, C.; et al. Cancer-associated fibroblasts induce sorafenib resistance of hepatocellular carcinoma cells through CXCL12/FOLR1. BMC Cancer 2023, 23, 1198. [Google Scholar] [CrossRef]
- Chang, K.; Chen, Y.; Zhang, X.; Zhang, W.; Xu, N.; Zeng, B.; Wang, Y.; Feng, T.; Dai, B.; Xu, F.; et al. DPP9 Stabilizes NRF2 to Suppress Ferroptosis and Induce Sorafenib Resistance in Clear Cell Renal Cell Carcinoma. Cancer Res. 2023, 83, 3940–3955. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, D.; Feng, J.; Jinsihan, D. MiR-600 mediates EZH2/RUNX3 signal axis to modulate breast cancer cell viability and sorafenib sensitivity. J. Biochem. Mol. Toxicol. 2024, 38, e23613. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Su, Z.F.; Yuan, Y. Synergistic effects of sorafenib in combination with gemcitabine or pemetrexed in lung cancer cell lines with K-ras mutations. Contemp. Oncol./Współczesna Onkol. 2016, 20, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, R.; Zhang, X.; Wu, F.; Li, S.; Yuan, Y. Combination treatment of gemcitabine and sorafenib exerts a synergistic inhibitory effect on non-small cell lung cancer in vitro and in vivo via the epithelial-to-mesenchymal transition process. Oncol. Lett. 2020, 20, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kutkowska, J.; Strzadala, L.; Rapak, A. Synergistic activity of sorafenib and betulinic acid against clonogenic activity of non-small cell lung cancer cells. Cancer Sci. 2017, 108, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Okuma, T.; Lorenzo, A.; Yokota, A.; Hino, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Miyazawa, K. Fingolimod sensitizes EGFR wild-type non-small cell lung cancer cells to lapatinib or sorafenib and induces cell cycle arrest. Oncol. Rep. 2019, 42, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ju, R.; Shi, J.; Chen, W.; Sun, F.; Zhu, L.; Li, J.; Zhang, D.; Ye, C.; Guo, L. Carboxyamidotriazole synergizes with sorafenib to combat non–small cell lung cancer through inhibition of NANOG and aggravation of apoptosis. J. Pharmacol. Exp. Ther. 2017, 362, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, S.; Chen, X.; Zhang, S.; Wang, Z.; Mei, Q. The novel PI3K inhibitor S1 synergizes with sorafenib in non-small cell lung cancer cells involving the Akt-S6 signaling. Investig. New Drugs 2019, 37, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, H.; Xu, X.; Liu, H.; Wu, C.; Zhao, L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol. Lett. 2020, 19, 323–333. [Google Scholar] [CrossRef]
- Kim, S.J.; Puranik, N.; Yadav, D.; Lee, P.C.W. Lipid nanocarrier-based drug delivery systems: Therapeutic advances in the treatment of lung cancer. Int. J. Nanomed. 2023, 18, 2659–2676. [Google Scholar] [CrossRef]
- Zhong, T.; Liu, X.; Li, H.; Zhang, J. Co-delivery of sorafenib and crizotinib encapsulated with polymeric nanoparticles for the treatment of in vivo lung cancer animal model. Drug Deliv. 2021, 28, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Kulkarni, N.S.; Farrales, P.; Kanabar, D.D.; Parvathaneni, V.; Kunda, N.K.; Muth, A.; Gupta, V. Sorafenib Loaded Inhalable Polymeric Nanocarriers against Non-Small Cell Lung Cancer. Pharm. Res. 2020, 37, 67. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Bothiraja, C.; Mali, A.; Kamble, R. Investigation of sorafenib tosylate loaded liposomal dry powder inhaler for the treatment of non-small cell lung cancer. Part. Sci. Technol. 2021, 39, 990–999. [Google Scholar] [CrossRef]
- He, C.; Ali, D.J.; Sun, B.; Sun, B.C.; Xiao, Z.D. Microvesicles: The functional mediators in sorafenib resistance. Cancer Drug Resist. 2022, 5, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, A.; Kadam, S.M.; Liu, L.; Kelly, L.E.; Rapp, C.M.; Chen, Y.; Sahu, R.P. Gemcitabine Induces Microvesicle Particle Release in a Platelet-Activating Factor-Receptor-Dependent Manner via Modulation of the MAPK Pathway in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.J.; Thyagarajan, A.; Chen, Y.; Travers, J.B.; Sahu, R.P. Platelet-activating factor- receptor signaling mediates targeted therapies-induced microvesicle particles release in lung cancer cells. Int. J. Mol. Sci. 2020, 21, 8517. [Google Scholar] [CrossRef]
- Chauhan, S.J.; Thyagarajan, A.; Sahu, R.P. Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 6772. [Google Scholar] [CrossRef]
- Hackler, P.C.; Reuss, S.; Konger, R.L.; Travers, J.B.; Sahu, R.P. Systemic Platelet-activating Factor Receptor Activation Augments Experimental Lung Tumor Growth and Metastasis. Cancer Growth Metastasis 2014, 7, 27–32. [Google Scholar] [CrossRef]
- Moore, C.; Kosgodage, U.; Lange, S.; Inal, J.M. The emerging role of exosome and microvesicle-(EMV-) based cancer therapeutics and immunotherapy. Int. J. Cancer 2017, 141, 428–436. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Saintigny, P.; Liu, S.; Kim, E.S.; Tsao, A.S.; Herbst, R.S.; Alden, C.; Lee, J.J.; Tang, X.; Stewart, D.J.; et al. Comprehensive biomarker analysis and final efficacy results of sorafenib in the BATTLE trial. Clin. Cancer Res. 2013, 19, 6967–6975. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Lee, J.J.; Wistuba, I.I.; Tsao, A.S.; Fossella, F.V.; Kalhor, N.; Gupta, S.; Byers, L.A.; Izzo, J.G.; Gettinger, S.N.; et al. The BATTLE-2 study: A biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3638–3647. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Hirsh, V.; Zhang, L.; de Marinis, F.; Yang, J.C.; Wakelee, H.A.; Seto, T.; Wu, Y.-L.; Novello, S.; Juhász, E.; et al. Monotherapy Administration of Sorafenib in Patients with Non-Small Cell Lung Cancer (MISSION) Trial: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib in Patients with Relapsed or Refractory Predominantly Nonsquamous Non-Small-Cell Lung Cancer after 2 or 3 Previous Treatment Regimens. J. Thorac. Oncol. 2015, 10, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, A.M.C.; Mellema, W.W.; Groen, H.J.; van Wijk, A.; Burgers, S.A.; Kunst, P.W.; Thunnissen, E.; Heideman, D.A.; Smit, E.F. A Phase II study of sorafenib in patients with platinum-pretreated, advanced (Stage IIIb or IV) non–small cell lung cancer with a KRAS mutation. Clin. Cancer Res. 2013, 19, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Rubin, M.S.; Gian, V.G.; Shipley, D.L.; Burris, H.A.; Kosloff, R.A.; Shih, K.C.; Quinn, R.; Greco, F.A.; Hainsworth, J.D. Sorafenib and continued erlotinib or sorafenib alone in patients with advanced non-small cell lung cancer progressing on erlotinib: A randomized phase II study of the Sarah Cannon Research Institute (SCRI). Lung Cancer 2017, 113, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Mattonet, C.; Scheffler, M.; Taubert, M.; Gardizi, M.; Sos, M.L.; Michels, S.; Fischer, R.N.; Limburg, M.; Abdulla, D.S.; et al. Sorafenib and everolimus in patients with advanced solid tumors and KRAS-mutated NSCLC: A phase I trial with early pharmacodynamic FDG-PET assessment. Cancer Med. 2020, 9, 4991–5007. [Google Scholar] [CrossRef]
- Zhang, J.; Gold, K.A.; Kim, E. Sorafenib in non-small cell lung cancer. Expert Opin. Investig. Drugs 2012, 21, 1417–1426. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Treatment(s) | Cell Line(s) | Drug Concentration | Combination Index (CI) |
|---|---|---|---|
| Sorafenib/gemcitabine [34] | A549 | SF 0–32 µM/ GEM 0–64 µM | 0.86 |
| Sorafenib/pemetrexed [34] | A549 | SF 0–32 µM/ PEM 0–32 µM | 0.63 |
| Sorafenib/gemcitabine [35] | A549 | SF 5–10 µM/ GEM 5–10 µM | 0.65 |
| Sorafenib/betulinic acid [36] | A549, H358, A429 | SF 1.3 µM/ BA 3 µM | 0.749, 0.802, 0.497 |
| Sorafenib/fingolimod [37] | A549 | SF 2.5–10 µM/ FTY 2.5–10 µM | 0.74 |
| Sorafenib/CAI [38] | LLC | SF 0.5–10 µM/ CAI 1–20 µM | <1 |
| Sorafenib/CAI [38] | A459 | SF 0.5–10 µM/ CAI 1–20 µM | <1 |
| Sorafenib/CAI [38] | H1975 | SF 0.5–2.5 µM/ CAI 1–5 µM | <1 |
| Sorafenib/CAI [38] | H1975 | SF 5–10 µM/ CAI 10–20 µM | >1 |
| Treatment(s) | Biomarker(s) | Median PFS | Median OS |
|---|---|---|---|
| Sorafenib [9,51] | Multiple/Not specific | 2.83 months | 8.48 months |
| Sorafenib [9,51] | EGFR FISH-negative | 3.35 months * | Not reported |
| Sorafenib [9,51] | EGFR FISH-positive | 1.84 months * | Not reported |
| High-conc Sorafenib Sensitivity Signature [9,51] | EGFR wild-type | 3.61 months * | Not reported |
| Low-conc sorafenib sensitivity signature [9,51] | EGFR wild-type | 1.84 months * | Not reported |
| Treatment(s) | Biomarker(s) | Median PFS | Median OS |
|---|---|---|---|
| Sorafenib [53] | EGFR-mutant | 2.7 months * | 13.9 months * |
| Placebo | EGFR-mutant | 1.4 months * | 6.5 months * |
| Sorafenib [53] | KRAS-mutant | 2.6 months * | 6.5 months |
| Placebo | KRAS-mutant | 1.7 months * | 5.1 months |
| Treatment(s) | Biomarker(s) | Median PFS | Median OS |
|---|---|---|---|
| Sorafenib [54] | KRAS-mutant | 2.3 months | 5.3 months |
| Sorafenib [55] | Multiple/Not specific | 1.9 months | 11.9 months |
| Sorafenib/Erlotinib [55] | Multiple/Not specific | 3.1 months | 8.9 months |
| Sorafenib/Everolimus [56] | KRAS-mutant | 3.25 months | 5.85 months |
| Treatment(s) | Biomarker(s) | Findings |
|---|---|---|
| Sorafenib [9,51] | EGFR wild-type vs. mutant | DCR is higher among patients with wild-type EGFR tumors vs mutant EGFR tumors. PFS not significant. |
| Sorafenib [9,51] | EGFR FISH-positive vs. FISH-negative | DCR and median PFS higher in EGFR FISH-negative tumors |
| Sorafenib sensitivity signature Low vs. high conc [9,51] | EGFR wild-type | PFS higher among pts treated with high-conc SSS. DCR not significant. |
| Sorafenib sensitivity signature [52] | KRAS mutation | Median PFS and OS not significant. |
| Sorafenib vs. placebo [53] | EGFR mutation | Median PFS and OS higher in patients with mutant EGFR who received sorafenib vs those with mutant EGFR who received placebo. |
| Sorafenib vs. placebo [53] | KRAS mutation | PFS increased in individuals treated with sorafenib. OS not significant. |
| Sorafenib [54] | KRAS mutation | The type of KRAS mutation did not significantly impact PFS or OS. |
| Sorafenib + Everolimus [56] | KRAS mutation | No significant correlation between PMR and PFS/OS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrixson, M.; Gladkiy, Y.; Thyagarajan, A.; Sahu, R.P. Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer. Med. Sci. 2024, 12, 20. https://doi.org/10.3390/medsci12020020
Hendrixson M, Gladkiy Y, Thyagarajan A, Sahu RP. Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer. Medical Sciences. 2024; 12(2):20. https://doi.org/10.3390/medsci12020020
Chicago/Turabian StyleHendrixson, Morgann, Yevgeniy Gladkiy, Anita Thyagarajan, and Ravi P. Sahu. 2024. "Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer" Medical Sciences 12, no. 2: 20. https://doi.org/10.3390/medsci12020020
APA StyleHendrixson, M., Gladkiy, Y., Thyagarajan, A., & Sahu, R. P. (2024). Efficacy of Sorafenib-Based Therapies for Non-Small Cell Lung Cancer. Medical Sciences, 12(2), 20. https://doi.org/10.3390/medsci12020020