Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma


Abstract
:1. Introduction
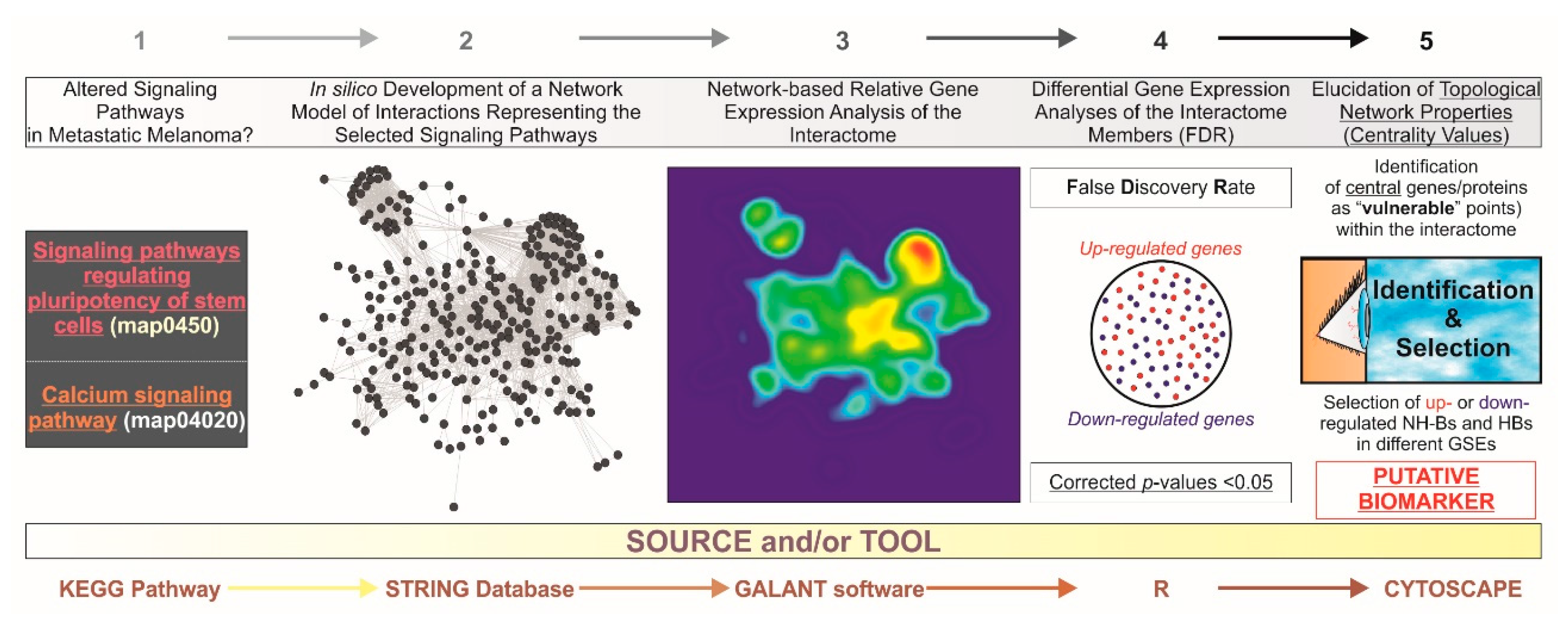
2. Material and Methods
2.1. Development of the Gene/Protein Interaction Network Model, Data Acquisition, and Processing
2.2. Relative Gene Expression Network Visualization and Microarray Analysis
2.3. Elucidation of the Topological Network Properties (Degree or Connectivity and Betweenness)
3. Results
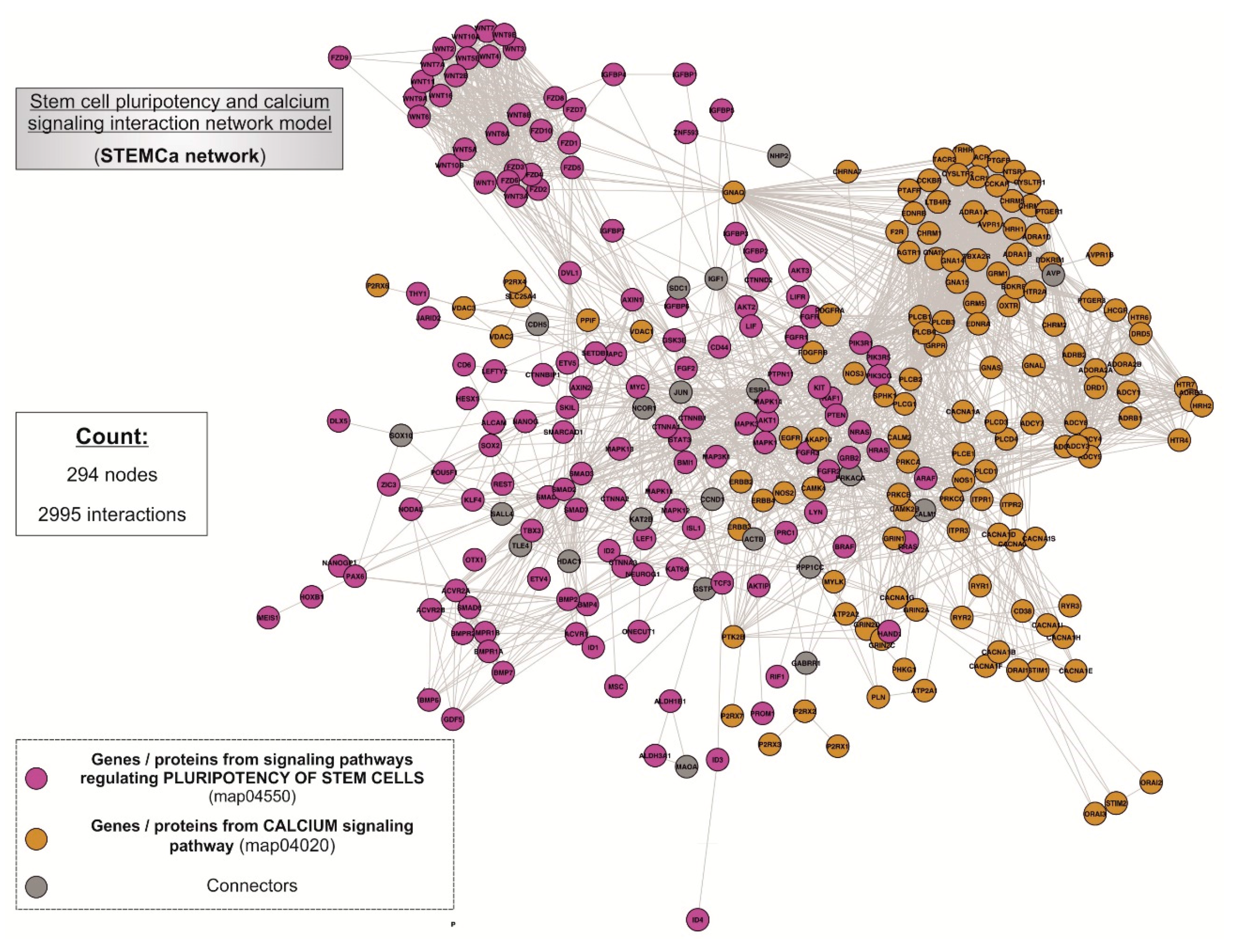
3.1. “STEMCa” Is an Integrative Network Model of Interactions for Stem Cell Pluripotency and Ca2+ Signaling Pathways
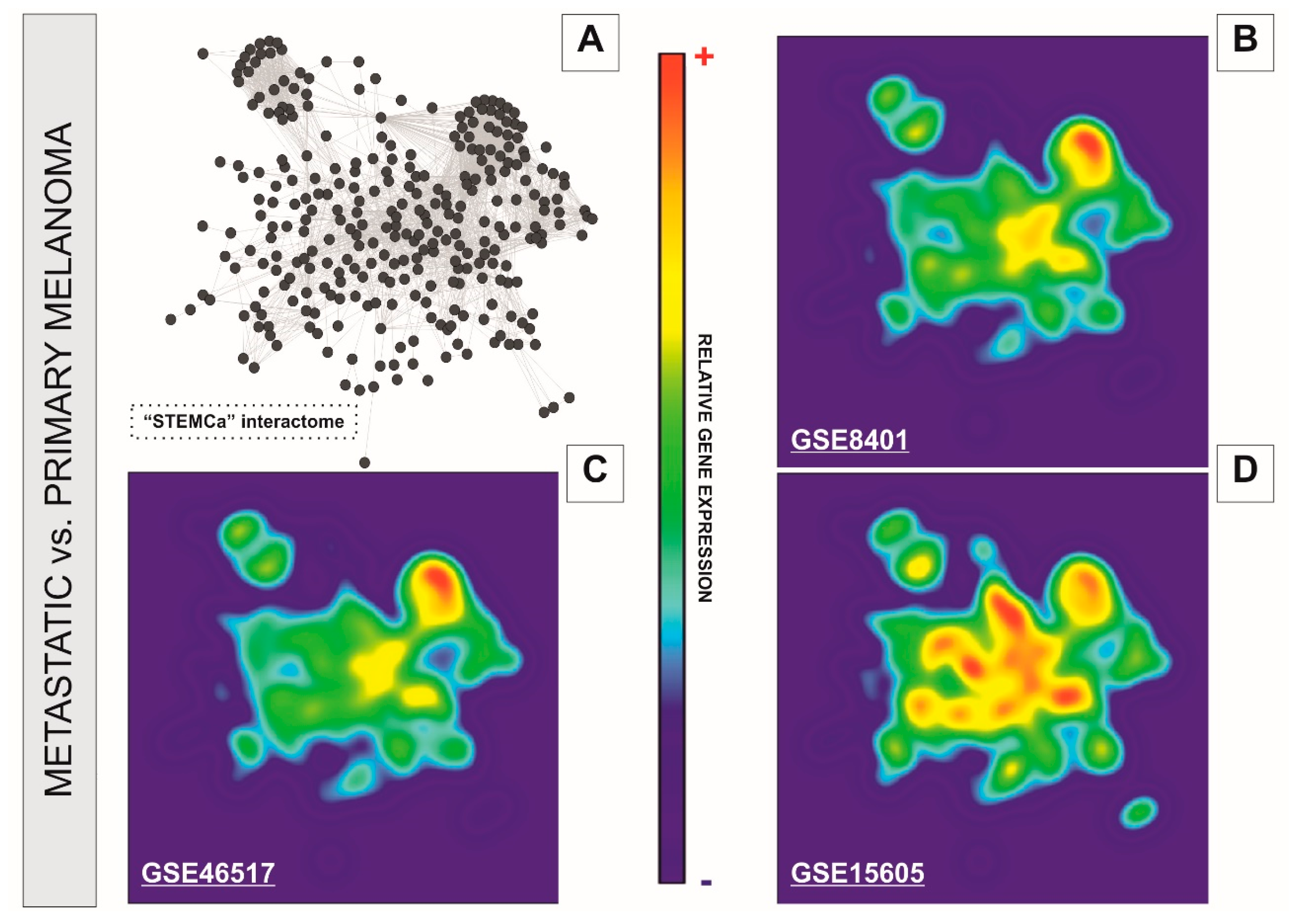
3.2. Network-Based Analyses of Stem Cell Pluripotency and Ca2+ Signaling Pathways Reveal Transcriptional Changes in Independent Datasets of Human Metastatic Melanoma
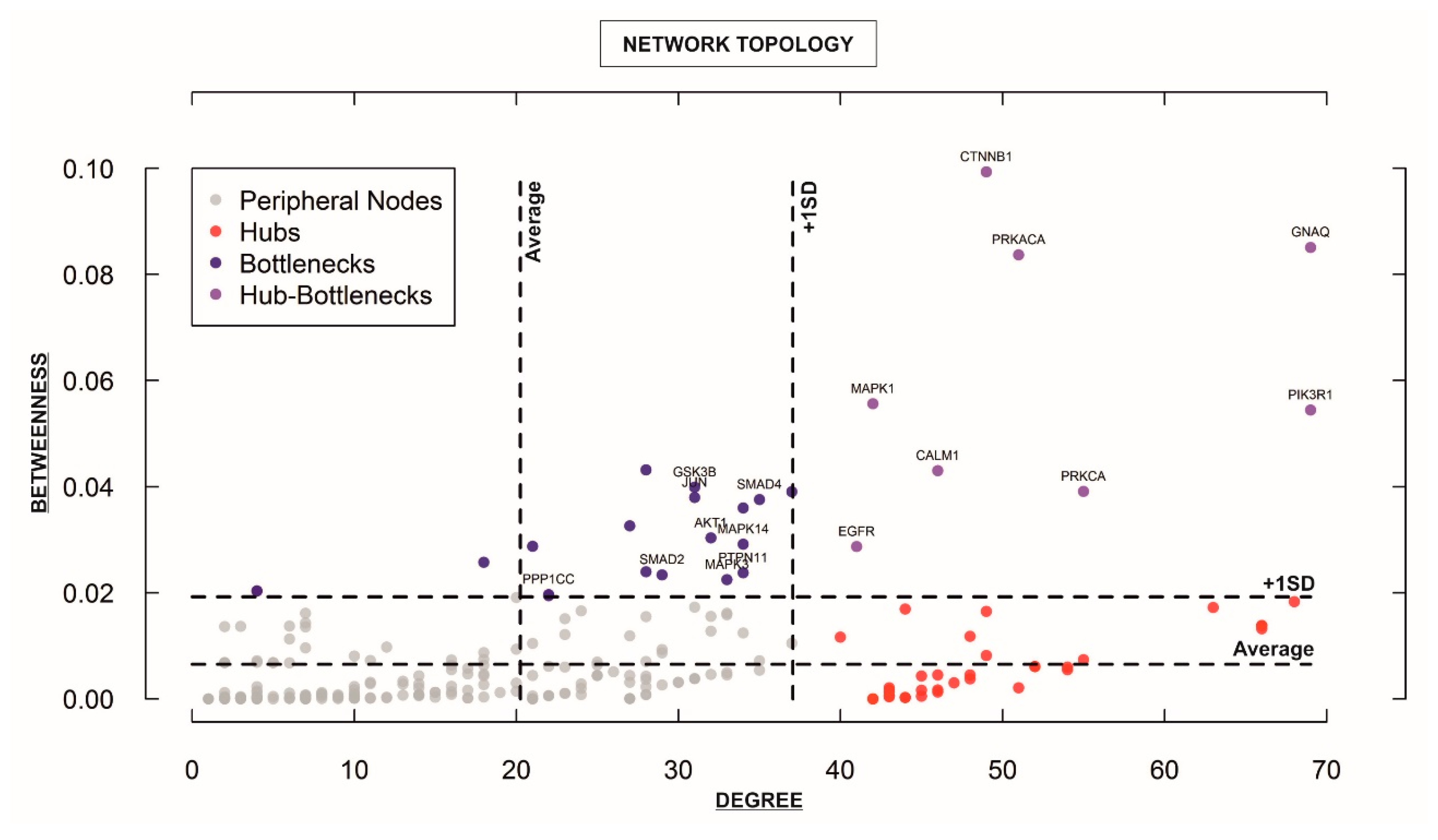
3.3. Elucidation of Key Hub Genes/Proteins within the Integrative “STEMCa” Interactome Suggests a Group of Candidate Biomarkers for Metastatic Melanoma
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koren, E.; Fuchs, Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist. Updat. 2012, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D. Cancer stem cells: Moving past the controversy. CNS Oncol. 2013, 2, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Malignant melanoma: Diagnosis, treatment and cancer stem cells. Neoplasma 2016, 63, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Noda, M. “Opening” the mesenchymal stem cell tool box. Eur. J. Dent. 2009, 3, 240–249. [Google Scholar] [PubMed]
- Dayanithi, G.; Verkhratsky, A. Calcium signaling in stem cells: Molecular physiology and multiple roles. Cell Calcium 2016, 59, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Kadio, B.; Yaya, S.; Basak, A.; Djè, K.; Gomes, J.; Mesenge, C. Calcium role in human carcinogenesis: A comprehensive analysis and critical review of literature. Cancer Metastasis Rev. 2016, 35, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Rizaner, N.; Onkal, R.; Fraser, S.P.; Pristerá, A.; Okuse, K.; Djamgoz, M.B. Intracellular calcium oscillations in strongly metastatic human breast and prostate cancer cells: Control by voltage-gated sodium channel activity. Eur. Biophys. J. 2016, 45, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Johann, D.J., Jr.; McGuigan, M.D.; Patel, A.R.; Tomov, S.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Fishman, D.A.; Whiteley, G.R.; Petricoin, E.F., III; et al. Clinical proteomics and biomarker discovery. Ann. N. Y. Acad. Sci. 2004, 1022, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Khunlertgit, N.; Yoon, B.J. Incorporating topological information for predicting robust cancer subnetwork markers in human protein-protein interaction network. BMC Bioinform. 2016, 17, 351. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Rybarczyk-Filho, J.L.; Gursoy, M.; Könönen, E.; Uitto, V.J.; Gursoy, U.V.; Cakmakci, L.; Moreira, J.C.; Gursoy, U.K. Bioinformatical and in vitro approaches to essential oil-induced matrix metalloproteinase inhibition. Pharm. Biol. 2012, 50, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Neves de Oliveira, B.H.; Gursoy, M.; Könönen, E.; Fonseca Moreira, J.C.; Gursoy, U.K.; Uitto, V.J. MMP-REDOX/NO interplay in periodontitis and its inhibition with Satureja hortensis L. essential oil. Chem. Biodivers. 2013, 10, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Gursoy, M.; de Oliveira, B.H.; Gelain, D.P.; Könönen, E.; Gursoy, U.K.; Moreira, J.C.; Uitto, V.J. Focussed microarray analysis of apoptosis in periodontitis and its potential pharmacological targeting by carvacrol. Arch. Oral Biol. 2014, 59, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Rybarczyk-Filho, J.L.; Salmina, A.B.; de Oliveira, B.H.; Noda, M.; Moreira, J.C. Exploring the multifactorial nature of autism through computational systems biology: Calcium and the Rho GTPase RAC1 under the spotlight. Neuromol. Med. 2013, 15, 364–383. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; de Oliveira, B.H.; Salmina, A.B.; Casanova, M.F.; Gelain, D.P.; Noda, M.; Verkhratsky, A.; Moreira, J.C. Altered expression of Alzheimer’s disease-related genes in the cerebellum of autistic patients: A model for disrupted brain connectome and therapy. Cell Death Dis. 2014, 5, e1250. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Gürsoy, M.; Neves de Oliveira, B.H.; Özdemir, V.; Könönen, E.; Gürsoy, U.K. A Systems Biology Approach to Reveal Putative Host-Derived Biomarkers of Periodontitis by Network Topology Characterization of MMP-REDOX/NO and Apoptosis Integrated Pathways. Front. Cell Infect. Microbiol. 2016, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; de Oliveira, B.H.; Casanova, M.F.; Casanova, E.L.; Noda, M.; Salmina, A.B.; Verkhratsky, A. Up-Regulation of Oligodendrocyte Lineage Markers in the Cerebellum of Autistic Patients: Evidence from Network Analysis of Gene Expression. Mol. Neurobiol. 2016, 53, 4019–4025. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shen, S.S.; Hoshida, Y.; Subramanian, A.; Ross, K.; Brunet, J.P.; Wagner, S.N.; Ramaswamy, S.; Mesirov, J.P.; Hynes, R.O. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol. Cancer Res. 2008, 6, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Kabbarah, O.; Nogueira, C.; Feng, B.; Nazarian, R.M.; Bosenberg, M.; Wu, M.; Scott, K.L.; Kwong, L.N.; Xiao, Y.; Cordon-Cardo, C.; et al. Integrative genome comparison of primary and metastatic melanomas. PLoS ONE 2010, 5, e10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raskin, L.; Fullen, D.R.; Giordano, T.J.; Thomas, D.G.; Frohm, M.L.; Cha, K.B.; Ahn, J.; Mukherjee, B.; Johnson, T.M.; Gruber, S.B. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Investig. Dermatol. 2013, 133, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Pawitan, Y.; Michiels, S.; Koscielny, S.; Gusnanto, A.; Ploner, A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics 2005, 21, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- Camilo, E.; Bovolenta, L.A.; Acencio, M.L.; Rybarczyk-Filho, J.L.; Castro, M.A.; Moreira, J.C.; Lemke, N. GALANT: A Cytoscape plugin for visualizing data as functional landscapes projected onto biological networks. Bioinformatics 2013, 29, 2505–2506. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massagué, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Talmadge, J.E. Evidence that intravenously derived murine pulmonary melanoma metastases can originate from the expansion of a single tumor cell. Cancer Res. 1986, 46, 5167–5171. [Google Scholar] [PubMed]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Apáti, Á.; Berecz, T.; Sarkadi, B. Calcium signaling in human pluripotent stem cells. Cell Calcium 2016, 59, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, O.; Forostyak, S.; Kortus, S.; Sykova, E.; Verkhratsky, A.; Dayanithi, G. Physiology of Ca2+ signaling in stem cells of different origins and differentiation stages. Cell Calcium 2016, 59, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Sonawane, M.D.; Song, K.S.; Kim, T. Biomarker detection technologies and future directions. Analyst 2016, 141, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.P.; Kuo, I.Y.; Alakus, H.; Frazer, K.A.; Harismendy, O.; Wang, Y.C.; Tseng, V.S. Network-based analysis identifies epigenetic biomarkers of esophageal squamous cell carcinoma progression. Bioinformatics 2014, 30, 3054–3061. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.O.; Henriques, J.P.; Bonatto, D. A systems pharmacology analysis of major chemotherapy combination regimens used in gastric cancer treatment: Predicting potential new protein targets and drugs. Curr. Cancer Drug Targets 2011, 11, 849–869. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Heynen, G.J.; Germano, G.; Willems, S.M.; Evers, B.; Vecchione, L.; Gambino, V.; Lieftink, C.; Beijersbergen, R.L.; Di Nicolantonio, F.; et al. PTPN11 Is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep. 2015, 12, 1978–1985. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Information | Topology | NH-B or HB | GSE8401 | GSE46517 | GSE15605 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | Ensemble ID | Sub. | Degree | Betweenness | Diff. Exp. | Corrected p-Value (FDR) | Diff. Exp. | Corrected p-Value (FDR) | Diff. Exp. | Corrected p-Value (FDR) | |
| AKT1 | ENSP00000270202 | ST | 32 | 0.03034247 | NH-B | --- | 0.073094859 | Down | 0.002572769 | --- | 0.99159187 |
| CALM1 | ENSP00000349467 | CON | 46 | 0.04297522 | HB | --- | 0.420640218 | Up | 0.014354422 | --- | 0.102753445 |
| CTNNB1 | ENSP00000344456 | ST | 49 | 0.09934346 | HB | Up | 0.000112883 | Up | 5.22113E−05 | --- | 0.134736908 |
| EGFR | ENSP00000275493 | Ca | 41 | 0.02867496 | HB | --- | 0.454469806 | Down | 4.09354E−18 | --- | 0.372953188 |
| ESR1 | ENSP00000206249 | CON | 37 | 0.03902762 | NH-B | --- | 0.34361801 | --- | 0.691913409 | --- | 0.858356832 |
| GABRR1 | ENSP00000412673 | CON | 4 | 0.02033756 | NH-B | --- | 0.516750581 | --- | 0.178807962 | Up | 0.030236148 |
| GNAQ | ENSP00000286548 | Ca | 69 | 0.08508797 | HB | Up | 0.014769785 | Up | 0.038122837 | --- | 0.174777485 |
| GSK3B | ENSP00000324806 | ST | 31 | 0.03985954 | NH-B | Down | 4.43231E−06 | Down | 2.75596E−10 | --- | 0.542667881 |
| GSTP1 | ENSP00000381607 | CON | 4 | 0.02033756 | NH-B | Down | 3.14081E−05 | Down | 2.61693E−06 | --- | 0.274434846 |
| HDAC1 | ENSP00000362649 | CON | 27 | 0.03262084 | NH-B | --- | 0.213434449 | --- | 0.710999269 | --- | 0.865172009 |
| IGF1 | ENSP00000302665 | CON | 28 | 0.04312205 | NH-B | --- | 0.697266013 | --- | 0.431465367 | --- | 0.66956775 |
| ITPR3 | ENSP00000363435 | Ca | 22 | 0.01942364 | NH-B | --- | 0.269623007 | --- | 0.671875922 | --- | 0.742562621 |
| JUN | ENSP00000360266 | CON | 31 | 0.03792962 | NH-B | Down | 8.92807E−06 | --- | 0.171366276 | --- | 0.900656682 |
| MAPK1 | ENSP00000215832 | ST | 42 | 0.05561423 | HB | Up | 0.000412172 | --- | 0.429091242 | --- | 0.608460569 |
| MAPK14 | ENSP00000229794 | ST | 34 | 0.02912488 | NH-B | --- | 0.605738821 | Up | 0.038642425 | --- | 0.358766287 |
| MAPK3 | ENSP00000263025 | ST | 33 | 0.02244545 | NH-B | Down | 1.10052E−05 | Down | 0.01074679 | --- | 0.999319565 |
| MYC | ENSP00000367207 | ST | 34 | 0.03598441 | NH-B | --- | 0.602836201 | --- | 0.274057104 | --- | 0.449615647 |
| NCOR1 | ENSP00000268712 | CON | 21 | 0.02875081 | NH-B | --- | 0.627351808 | --- | 0.257849878 | --- | 0.148831132 |
| PIK3R1 | ENSP00000274335 | ST | 69 | 0.05441236 | HB | --- | 0.383906559 | Down | 0.030059149 | --- | 0.057825554 |
| PPP1CC | ENSP00000335084 | CON | 22 | 0.01965509 | NH-B | Up | 1.52233E−05 | Up | 3.22551E−05 | --- | 0.307997432 |
| PRKACA | ENSP00000309591 | CON | 51 | 0.0837037 | HB | Down | 0.00193898 | Down | 0.006166758 | --- | 0.056947292 |
| PRKCA | ENSP00000408695 | Ca | 55 | 0.03906664 | HB | Up | 0.003860994 | --- | 0.227826787 | --- | 0.934220679 |
| PTPN11 | ENSP00000340944 | ST | 34 | 0.02374219 | NH-B | Up | 0.039908343 | Up | 0.009092992 | Up | 0.048264037 |
| SMAD2 | ENSP00000262160 | ST | 29 | 0.02336588 | NH-B | Up | 0.009466433 | --- | 0.924719732 | --- | 0.23296179 |
| SMAD3 | ENSP00000332973 | ST | 28 | 0.02392531 | NH-B | --- | 0.975647588 | --- | 0.09794078 | --- | 0.219862918 |
| SMAD4 | ENSP00000341551 | ST | 35 | 0.03753624 | NH-B | Up | 5.56315E−06 | Up | 0.000292094 | --- | 0.141417242 |
| TCF3 | ENSP00000262965 | ST | 18 | 0.02573384 | NH-B | --- | 0.803481248 | --- | 0.217139092 | --- | 0.260191349 |
| Mean | 20.37414 | 6.56E−03 | |||||||||
| SD | 16.80553 | 0.01276070 | |||||||||
| 1 SD | 37.17968 | 0.01932203 | |||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves de Oliveira, B.-H.; Dalmaz, C.; Zeidán-Chuliá, F. Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma. Med. Sci. 2018, 6, 23. https://doi.org/10.3390/medsci6010023
Neves de Oliveira B-H, Dalmaz C, Zeidán-Chuliá F. Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma. Medical Sciences. 2018; 6(1):23. https://doi.org/10.3390/medsci6010023
Chicago/Turabian StyleNeves de Oliveira, Ben-Hur, Carla Dalmaz, and Fares Zeidán-Chuliá. 2018. "Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma" Medical Sciences 6, no. 1: 23. https://doi.org/10.3390/medsci6010023
APA StyleNeves de Oliveira, B. -H., Dalmaz, C., & Zeidán-Chuliá, F. (2018). Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma. Medical Sciences, 6(1), 23. https://doi.org/10.3390/medsci6010023