An Overview of Dynamic Heterogeneous Oxidations in the Troposphere



Abstract
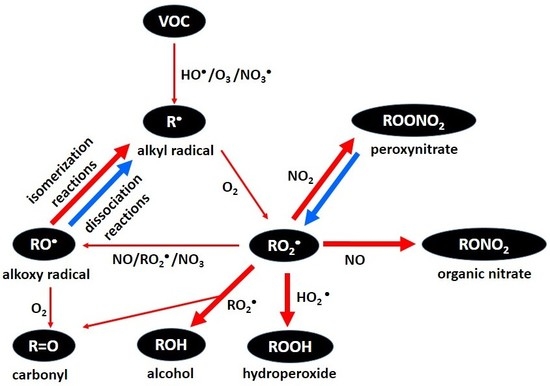
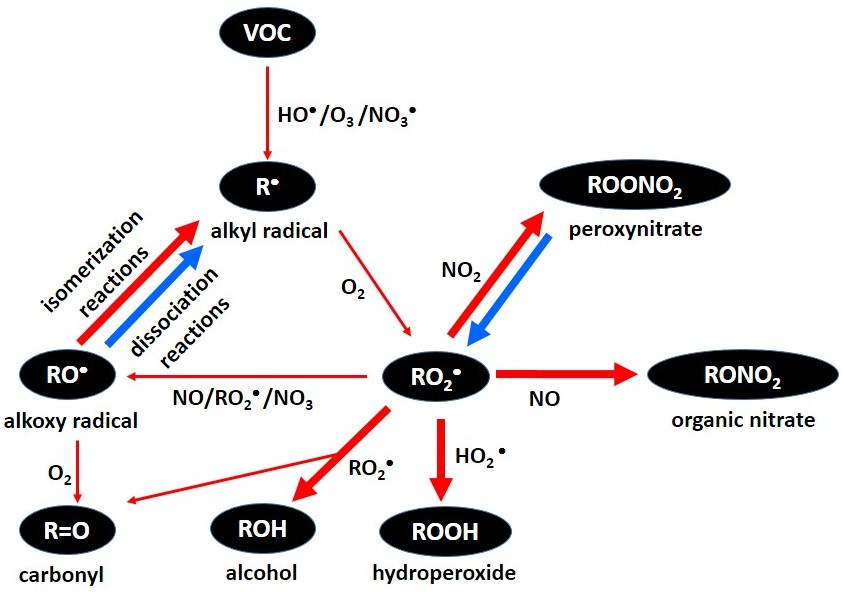
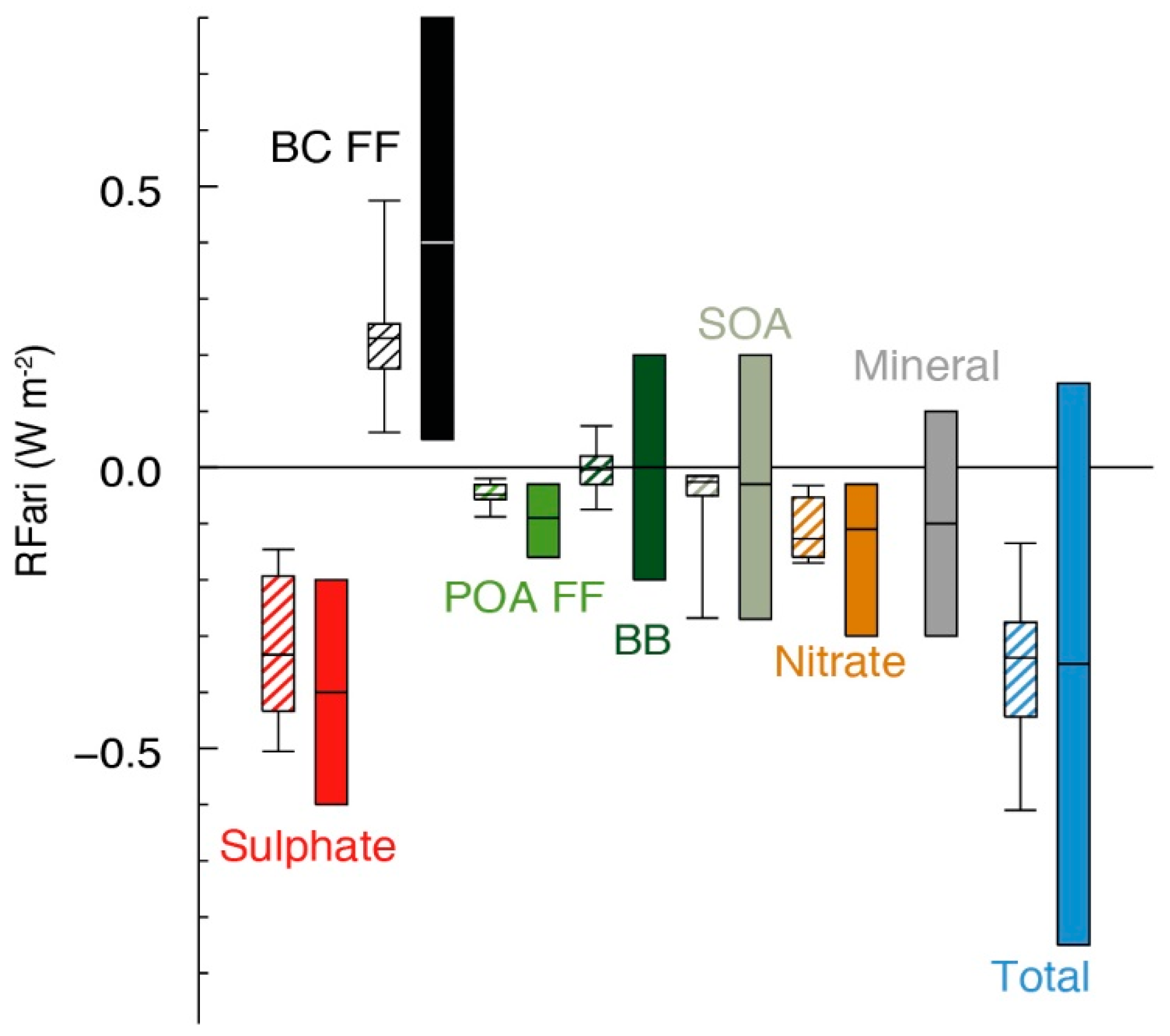
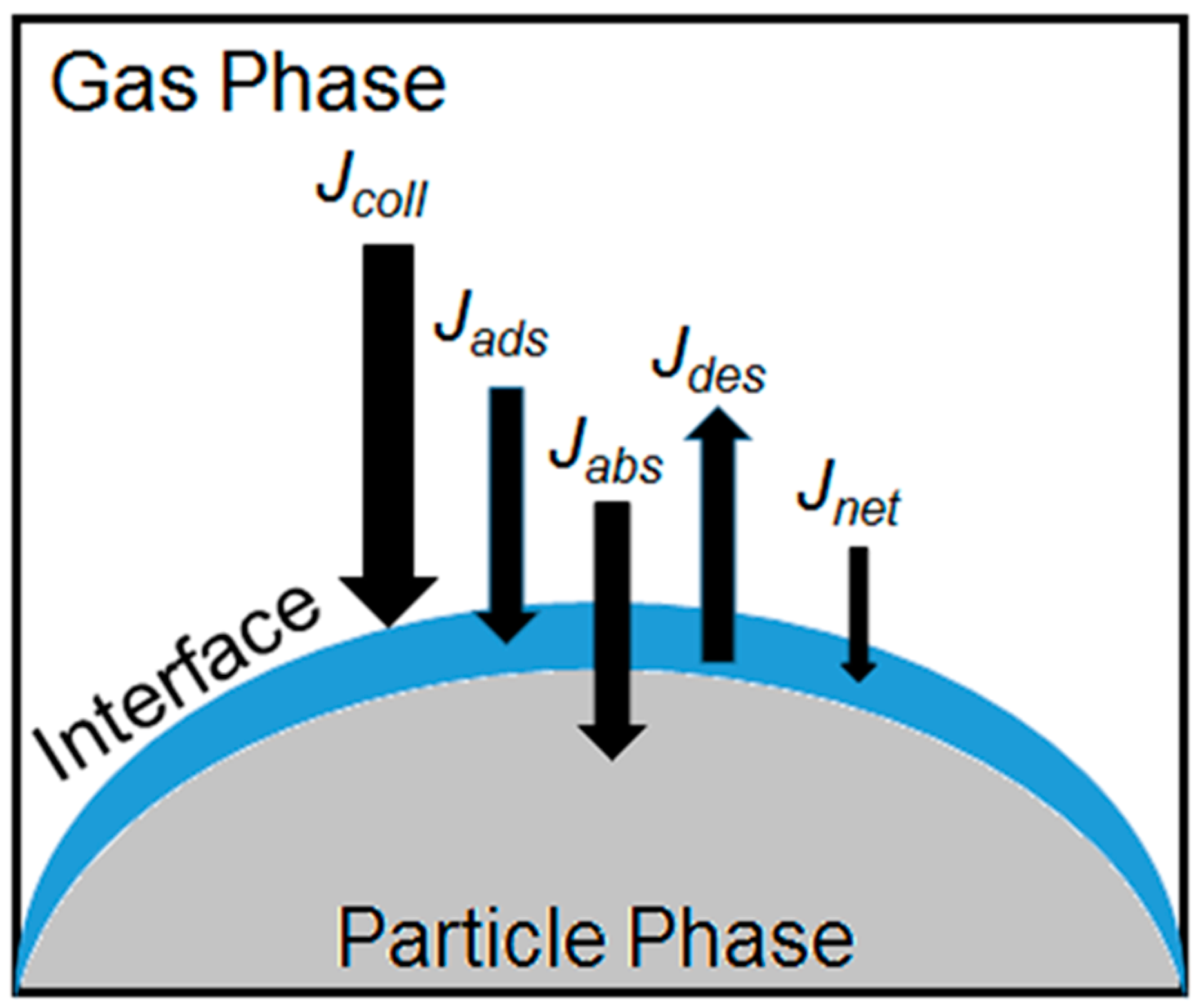
:
{kind=link}
{kind=link}
{kind=link}
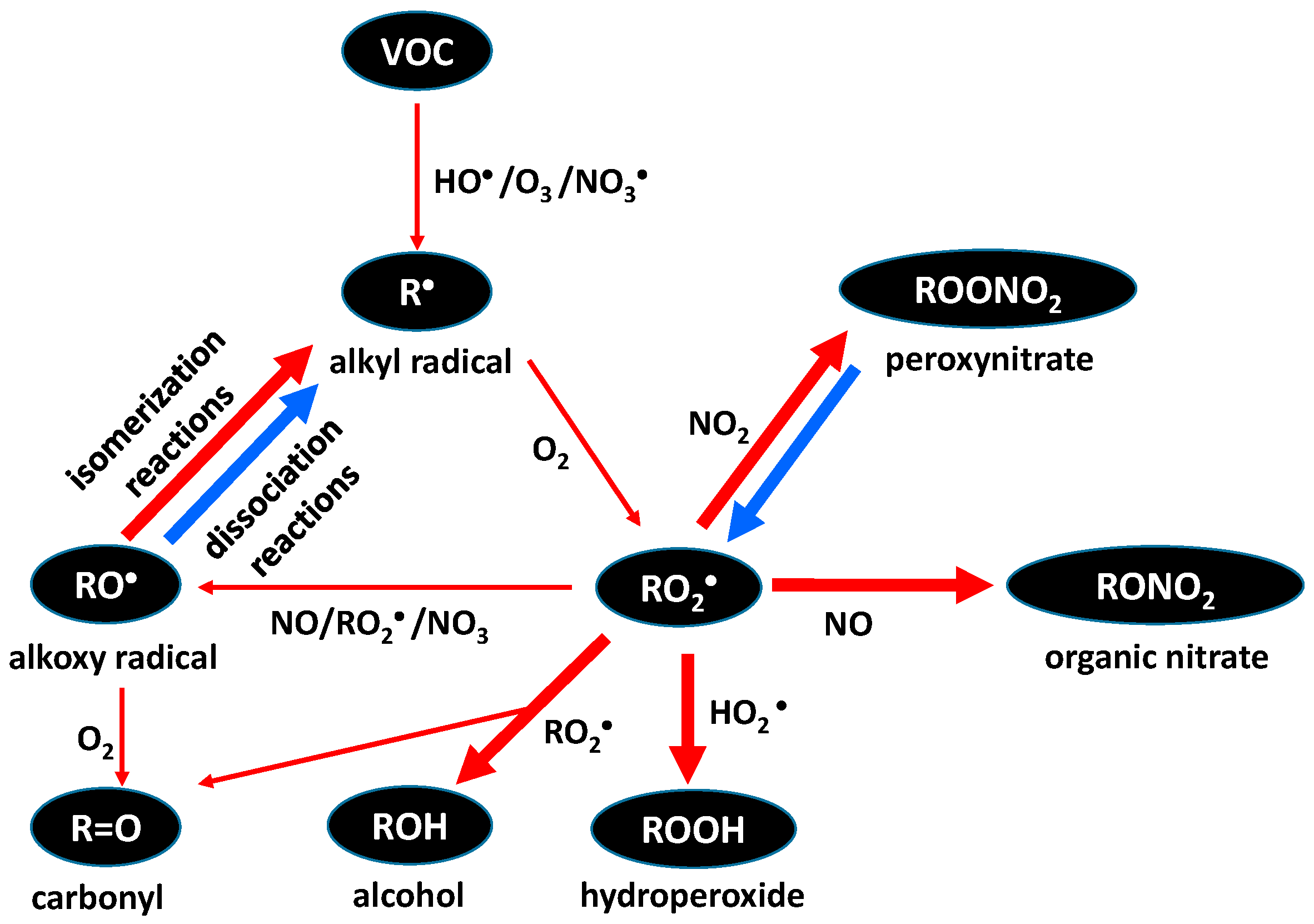{kind=link}
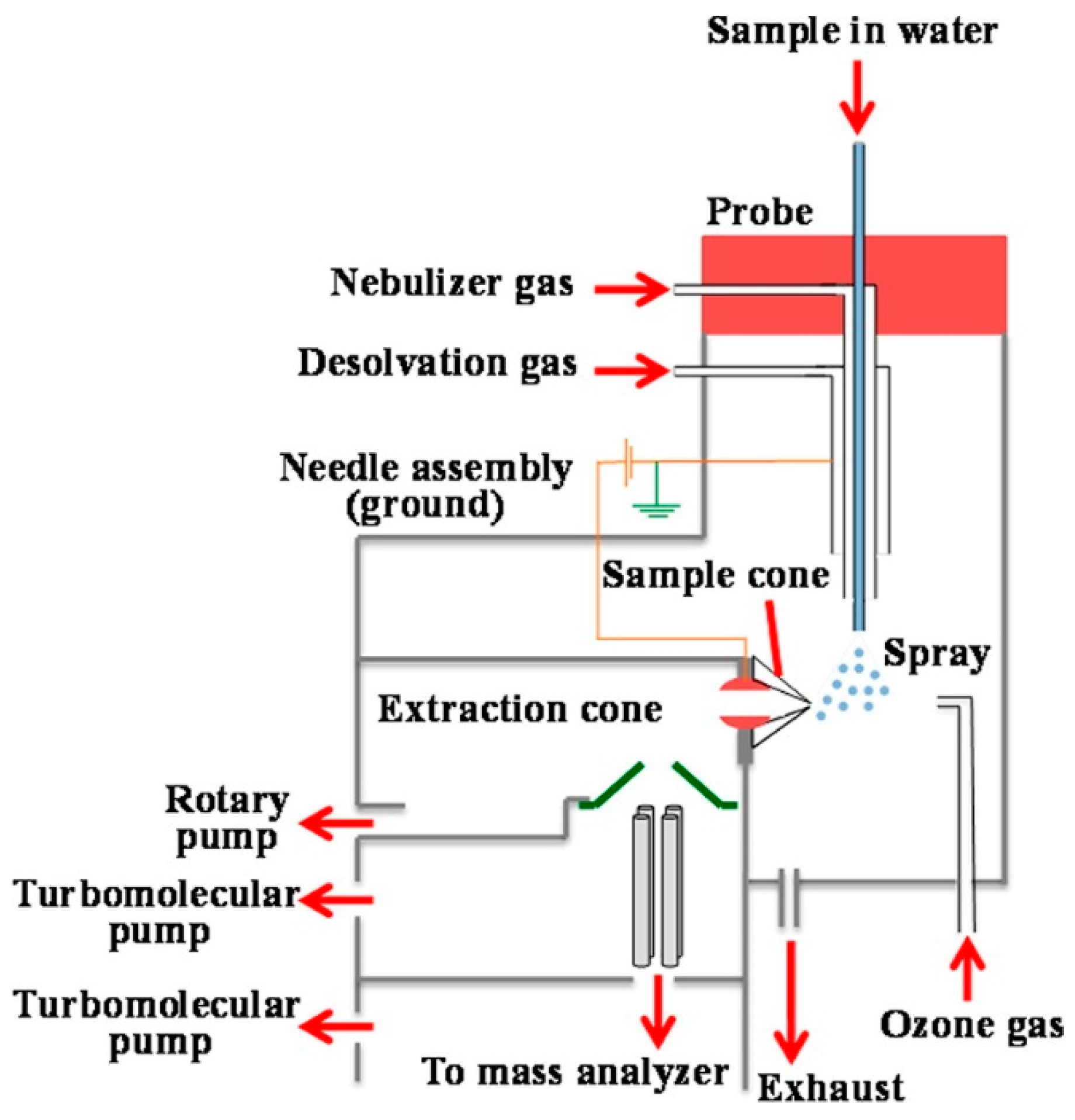{kind=link}
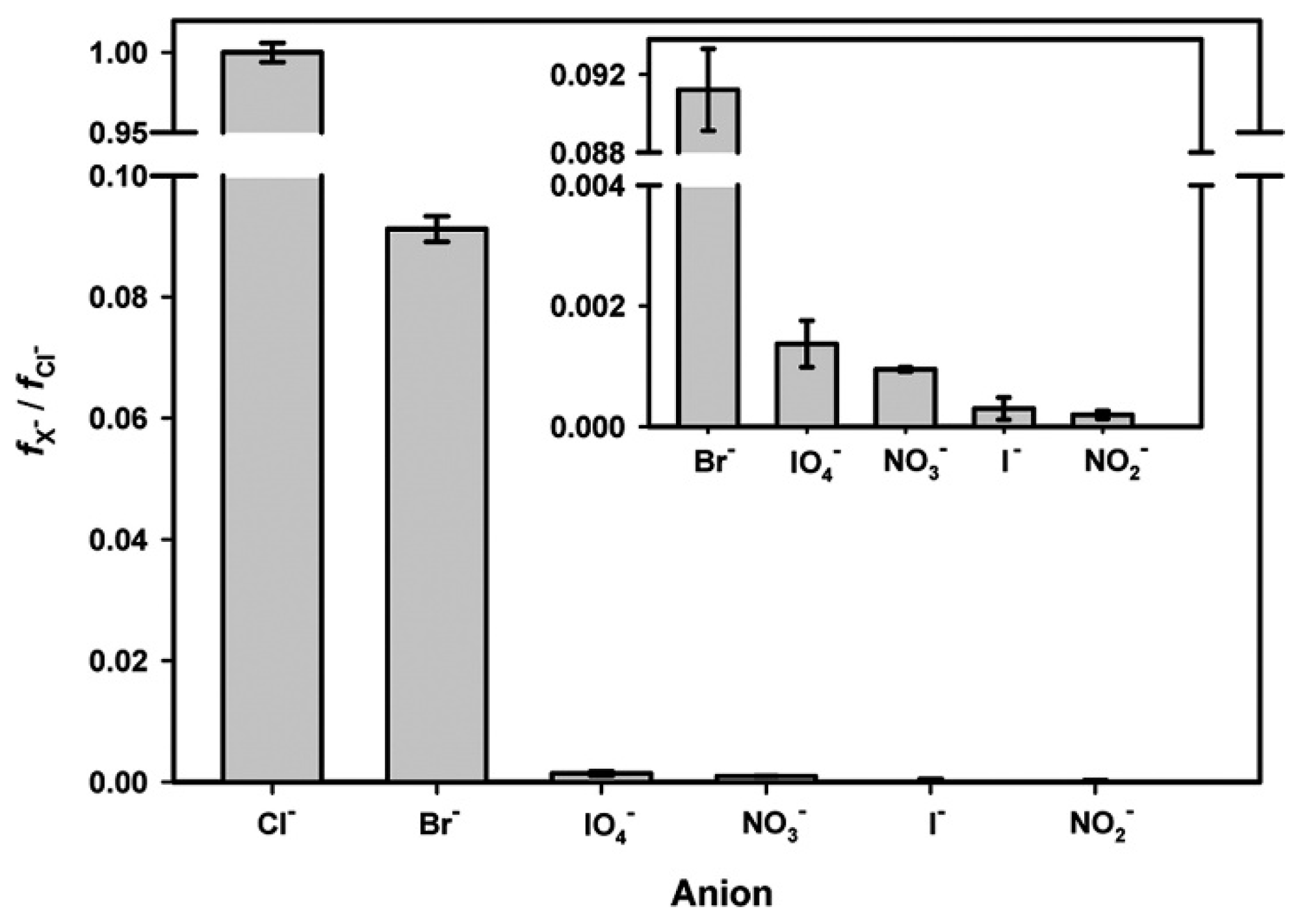{kind=link}
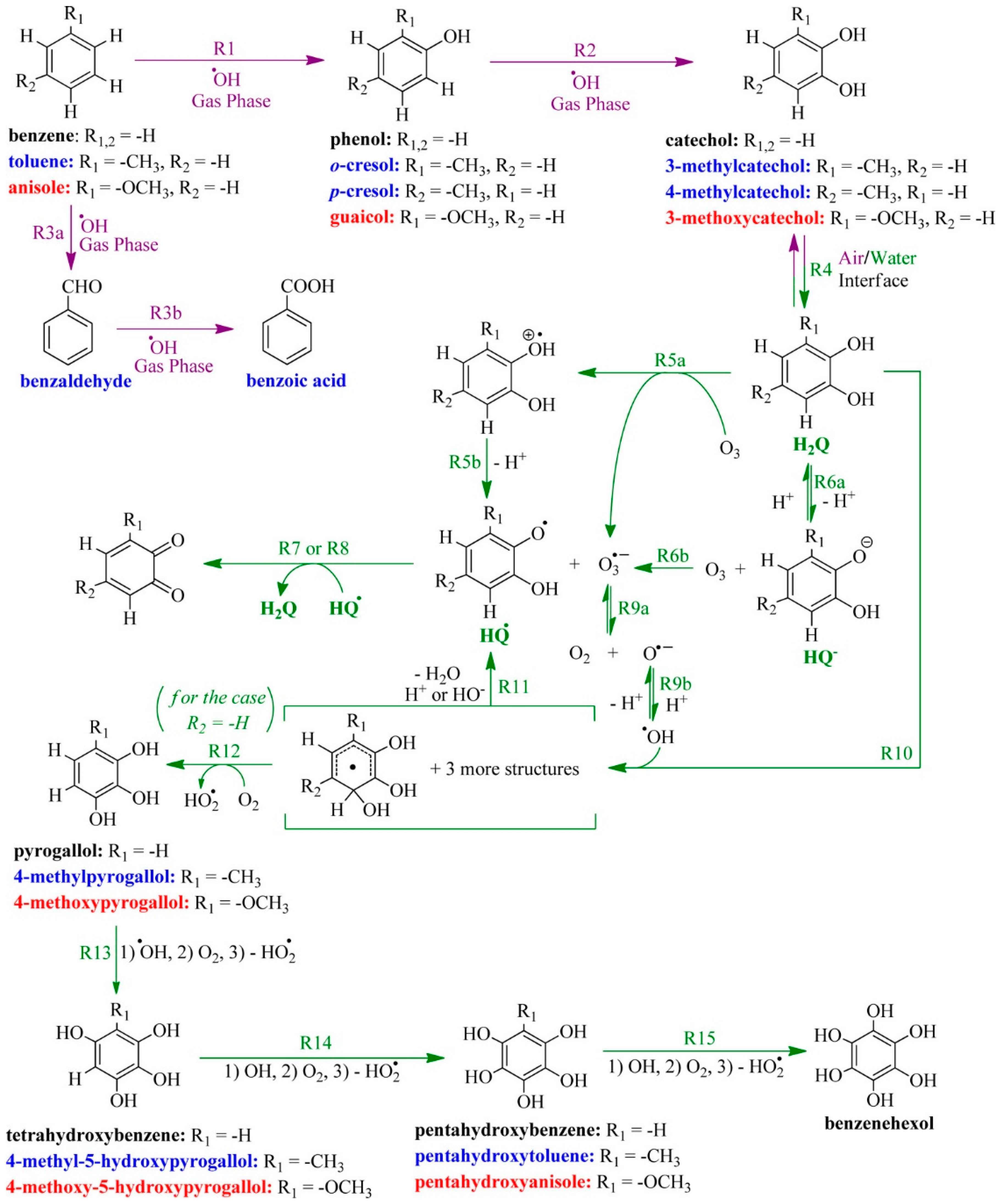{kind=link}
{kind=link}
1. Atmospheric Aerosols: Classification and Importance to Societies
2. The Effect of Anthropogenic Aerosols on the Earth’s Radiative Balance
3. Processing at Atmospheric Interfaces
4. Aging of Aerosols in the Atmosphere
5. Environmental Information from Laboratory Studies of Heterogeneous Reactions
6. Outcomes Learned from Recent Interfacial Studies
Author Contributions
Funding
Conflicts of Interest
References
- Von Schneidemesser, E.; Monks, P.S.; Allan, J.D.; Bruhwiler, L.; Forster, P.; Fowler, D.; Lauer, A.; Morgan, W.T.; Paasonen, P.; Righi, M.; et al. Chemistry and the linkages between air quality and climate change. Chem. Rev. 2015, 115, 3856–3897. [Google Scholar] [CrossRef] [PubMed]
- Andreae, M.O. Aerosols before pollution. Science 2007, 315, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Woodhouse, M.T.; Mann, G.W.; Carslaw, K.S.; Boucher, O. Sensitivity of cloud condensation nuclei to regional changes in dimethyl-sulphide emissions. Atmos. Chem. Phys. 2013, 13, 2723–2733. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.; Park, R.J.; Jacob, D.J.; Li, Q.B.; Yantosca, R.M.; Savarino, J.; Lee, C.C.W.; Thiemens, M.H. Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes. J. Geophys. Res. Atmos. 2005, 110. [Google Scholar] [CrossRef] [Green Version]
- Setyan, A.; Song, C.; Merkel, M.; Knighton, W.B.; Onasch, T.B.; Canagaratna, M.R.; Worsnop, D.R.; Wiedensohler, A.; Shilling, J.E.; Zhang, Q. Chemistry of new particle growth in mixed urban and biogenic emissions—Insights from CARES. Atmos. Chem. Phys. 2014, 14, 6477–6494. [Google Scholar] [CrossRef]
- Paasonen, P.; Asmi, A.; Petaja, T.; Kajos, M.K.; Aijala, M.; Junninen, H.; Holst, T.; Abbatt, J.P.D.; Arneth, A.; Birmili, W.; et al. Warming-induced increase in aerosol number concentration likely to moderate climate change. Nat. Geosci. 2013, 6, 438–442. [Google Scholar] [CrossRef]
- Wu, S.; Mickley, L.J.; Jacob, D.J.; Rind, D.; Streets, D.G. Effects of 2000–2050 changes in climate and emissions on global tropospheric ozone and the policy-relevant background surface ozone in the United States. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef] [Green Version]
- Ammann, M.; Klimont, Z.; Wagner, F. Regional and global emissions of air pollutants: Recent trends and future scenarios. Annu. Rev. Environ. Resour. 2013, 38, 31–55. [Google Scholar] [CrossRef]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P.I.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.G.; Jones, C.D.; Collins, W.J.; Johnson, C.E.; Derwent, R.G. Effect of climate change on isoprene emissions and surface ozone levels. Geophys. Res. Lett. 2003, 30. [Google Scholar] [CrossRef]
- Wiedinmyer, C.; Tie, X.; Guenther, A.; Neilson, R.; Granier, C. Future changes in biogenic isoprene emissions: How might they affect regional and global atmospheric chemistry? Earth Interact. 2006, 10, 1–19. [Google Scholar] [CrossRef]
- Lathière, J.; Hauglustaine, D.A.; Friend, A.D.; De Noblet-Ducoudré, N.; Viovy, N.; Folberth, G.A. Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos. Chem. Phys. 2006, 6, 2129–2146. [Google Scholar] [CrossRef] [Green Version]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.-M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 571–658. [Google Scholar]
- Wang, Y.; Shen, L.; Wu, S.; Mickley, L.; He, J.; Hao, J. Sensitivity of surface ozone over China to 2000–2050 global changes of climate and emissions. Atmos. Environ. 2013, 75, 374–382. [Google Scholar] [CrossRef]
- Doherty, R.M.; Wild, O.; Shindell, D.T.; Zeng, G.; MacKenzie, I.A.; Collins, W.J.; Fiore, A.M.; Stevenson, D.S.; Dentener, F.J.; Schultz, M.G.; et al. Impacts of climate change on surface ozone and intercontinental ozone pollution: A multi-model study. J. Geophys. Res. Atmos. 2013, 118, 3744–3763. [Google Scholar] [CrossRef] [Green Version]
- Naik, V.; Horowitz, L.W.; Fiore, A.M. Impact of preindustrial to present-day changes in short-lived pollutant emissions on atmospheric composition and climate forcing. J. Geophys. Res. Atmos. 2013, 118, 8086–8110. [Google Scholar] [CrossRef] [Green Version]
- Levy, H.; Horowitz, L.W.; Schwarzkopf, M.D.; Ming, Y.; Golaz, J.-C.; Naik, V.; Ramaswamy, V. The roles of aerosol direct and indirect effects in past and future climate change. J. Geophys. Res. Atmos. 2013, 118, 4521–4532. [Google Scholar] [CrossRef] [Green Version]
- Rotstayn, L.D.; Collier, M.A.; Chrastansky, A.; Jeffrey, S.J.; Luo, J.J. Projected effects of declining aerosols in RCP4.5: Unmasking global warming? Atmos. Chem. Phys. 2013, 13, 10883–10905. [Google Scholar] [CrossRef]
- Whitby, K.T. The physical characteristics of sulfur aerosols. Atmos. Environ. 1978, 12, 135–159. [Google Scholar] [CrossRef]
- Conte, M.; Donateo, A.; Dinoi, A.; Belosi, F.; Contini, D. Case study of particle number fluxes and size distributions during nucleation events in southeastern italy in the summer. Atmosphere 2015, 6, 942–959. [Google Scholar] [CrossRef]
- Hussein, T.; Martikainen, J.; Junninen, H.; Sogacheva, L.; Wagner, R.; Maso, M.D.; Riipinen, I.; Aalto, P.P.; Kulmala, M. Observation of regional new particle formation in the urban atmosphere. Tellus B 2008, 60, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Aalto, P.; Hämeri, K.; Becker, E.; Weber, R.; Salm, J.; Mäkelä, J.M.; Hoell, C.; O’dowd, C.D.; Hansson, H.-C.; Väkevä, M.; et al. Physical characterization of aerosol particles during nucleation events. Tellus B 2001, 53, 344–358. [Google Scholar] [CrossRef]
- Williams, J.; de Reus, M.; Krejci, R.; Fischer, H.; Ström, J. Application of the variability-size relationship to atmospheric aerosol studies: Estimating aerosol lifetimes and ages. Atmos. Chem. Phys. 2002, 2, 133–145. [Google Scholar] [CrossRef]
- Petroff, A.; Zhang, L. Development and validation of a size-resolved particle dry deposition scheme for application in aerosol transport models. Geosci. Model Dev. 2010, 3, 753–769. [Google Scholar] [CrossRef] [Green Version]
- Pryor, S.; Binkowski, F. An analysis of the time scales associated with aerosol processes during dry deposition. Aerosol. Sci. Technol. 2004, 38, 1091–1098. [Google Scholar] [CrossRef]
- Jacobs, D.J. Introduction to Atmospheric Chemistry; Princeton University Press: Princeton, NJ, USA, 1999. [Google Scholar]
- Monks, P.S.; Archibald, A.T.; Colette, A.; Cooper, O.; Coyle, M.; Derwent, R.; Fowler, D.; Granier, C.; Law, K.S.; Mills, G.E.; et al. Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer. Atmos. Chem. Phys. 2015, 15, 8889–8973. [Google Scholar] [CrossRef] [Green Version]
- Brook, R.D.; Rajagopalan, S. Particulate matter, air pollution, and blood pressure. J. Am. Soc. Hypertens. 2009, 3, 332–350. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Burnett, R.T.; Thun, M.J. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J. Am. Med. Assoc. 2002, 287, 1132–1141. [Google Scholar] [CrossRef]
- WHO. Review of Evidence on Health Aspects of Air Pollution (REVIHAAP) Project; WHO: Copenhagen, Denmark, 2013. [Google Scholar]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Tuomisto, J.T.; Wilson, A.; Evans, J.S.; Tainio, M. Uncertainty in mortality response to airborne fine particulate matter: Combining European air pollution experts. Reliab. Eng. Syst. Saf. 2008, 93, 732–744. [Google Scholar] [CrossRef]
- Lippmann, M.; Chen, L.C.; Gordon, T.; Ito, K.; Thurston, G.D. National Particle Component Toxicity (NPACT) Initiative: Integrated epidemiologic and toxicologic studies of the health effects of particulate matter components. Res. Rep. Health Eff. Inst. 2013, 177, 5–13. [Google Scholar]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Myhre, G.; Samset, B.H.; Schulz, M.; Balkanski, Y.; Bauer, S.; Berntsen, T.K.; Bian, H.; Bellouin, N.; Chin, M.; Diehl, T.; et al. Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations. Atmos. Chem. Phys. 2013, 13, 1853–1877. [Google Scholar] [CrossRef]
- Schuyler, T.J.; Guzman, M.I. Unmanned aerial systems for monitoring trace tropospheric gases. Atmosphere 2017, 8, 206. [Google Scholar] [CrossRef]
- Petters, M.D.; Kreidenweis, S.M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Duplissy, J.; DeCarlo, P.F.; Dommen, J.; Alfarra, M.R.; Metzger, A.; Barmpadimos, I.; Prevot, A.S.H.; Weingartner, E.; Tritscher, T.; Gysel, M.; et al. Relating hygroscopicity and composition of organic aerosol particulate matter. Atmos. Chem. Phys. 2011, 11, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Scheuer, E.; Dibb, J.; Ziemba, L.D.; Thornhill, K.L.; Anderson, B.E.; Wisthaler, A.; Mikoviny, T.; Devi, J.J.; Bergin, M.; et al. Brown carbon in the continental troposphere. Geophys. Res. Lett. 2014, 41, 2191–2195. [Google Scholar] [CrossRef] [Green Version]
- Twomey, S. The influence of pollution on the shortwave albedo of clouds. J. Atmos. Sci. 1977, 34, 1149–1152. [Google Scholar] [CrossRef]
- Isaksen, I.S.A.; Granier, C.; Myhre, G.; Berntsen, T.K.; Dalsøren, S.B.; Gauss, M.; Klimont, Z.; Benestad, R.; Bousquet, P.; Collins, W.; et al. Atmospheric composition change: Climate-chemistry interactions. Atmos. Environ. 2009, 43, 5138–5192. [Google Scholar] [CrossRef]
- Albrecht, B.A. Aerosols, cloud microphysics, and fractional cloudiness. Science 1989, 245, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z. Estimation of cloud condensation nuclei concentration from aerosol optical quantities: Influential factors and uncertainties. Atmos. Chem. Phys. 2014, 14, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Kravitz, B.; Wang, H.; Rasch, P.J.; Morrison, H.; Solomon, A.B. Process-model simulations of cloud albedo enhancement by aerosols in the Arctic. Philos. Trans. R. Soc. A 2014, 372. [Google Scholar] [CrossRef] [PubMed]
- Usher, C.R.; Michel, A.E.; Grassian, V.H. Reactions on mineral dust. Chem. Rev. 2003, 103, 4883–4940. [Google Scholar] [CrossRef] [PubMed]
- Sokolik, I.N.; Winker, D.M.; Bergametti, G.; Gillette, D.A.; Carmichael, G.; Kaufman, Y.J.; Gomes, L.; Schuetz, L.; Penner, J.E. Introduction to special section: Outstanding problems in quantifying the radiative impacts of mineral dust. J. Geophys. Res. Atmos. 2001, 106, 18015–18027. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, V.; Carmichael, G. Global and regional climate changes due to black carbon. Nat. Geosci. 2008, 1, 221–227. [Google Scholar] [CrossRef]
- Flanner, M.G. Arctic climate sensitivity to local black carbon. J. Geophys. Res. Atmos. 2013, 118, 1840–1851. [Google Scholar] [CrossRef] [Green Version]
- AMAP. The Impact of Black Carbon on the Arctic Climate; Arctic Monitoring and Assessment Programme (AMAP): Oslo, Norway, 2011. [Google Scholar]
- Johnson, D.; Utembe, S.R.; Jenkin, M.E.; Derwent, R.G.; Hayman, G.D.; Alfarra, M.R.; Coe, H.; McFiggans, G. Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK. Atmos. Chem. Phys. 2006, 6, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.L.; Donahue, N.M.; Shrivastava, M.K.; Weitkamp, E.A.; Sage, A.M.; Grieshop, A.P.; Lane, T.E.; Pierce, J.R.; Pandis, S.N. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science 2007, 315, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Heald, C.L.; Jacob, D.J.; Park, R.J.; Russell, L.M.; Huebert, B.J.; Seinfeld, J.H.; Liao, H.; Weber, R.J. A large organic aerosol source in the free troposphere missing from current models. Geophys. Res. Lett. 2005, 32. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J.W.; Heil, A.; Andreae, M.O.; Benedetti, A.; Chubarova, N.; Jones, L.; Morcrette, J.J.; Razinger, M.; Schultz, M.G.; Suttie, M.; et al. Biomass burning emissions estimated with a global fire assimilation system based on observed fire radiative power. Biogeoscience 2012, 9, 527–554. [Google Scholar] [CrossRef] [Green Version]
- Tosca, M.G.; Randerson, J.T.; Zender, C.S. Global impact of smoke aerosols from landscape fires on climate and the Hadley circulation. Atmos. Chem. Phys. 2013, 13, 5227–5241. [Google Scholar] [CrossRef]
- Moise, T.; Flores, J.M.; Rudich, Y. Optical properties of secondary organic aerosols and their changes by chemical processes. Chem. Rev. 2015, 115, 4400–4439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, G.; Guo, S.; Zamora, M.L.; Ying, Q.; Lin, Y.; Wang, W.; Hu, M.; Wang, Y. Formation of Urban Fine Particulate Matter. Chem. Rev. 2015, 115, 3803–3855. [Google Scholar] [CrossRef] [PubMed]
- Pusede, S.E.; Steiner, A.L.; Cohen, R.C. Temperature and recent trends in the chemistry of continental surface ozone. Chem. Rev. 2015, 115, 3898–3918. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Camm, R.C.; Guzman, M.I. Catechol oxidation by ozone and hydroxyl radicals at the air–water interface. Environ. Sci. Technol. 2014, 48, 14352–14360. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Zhou, R.; Guzman, M.I. Heterogeneous oxidation of catechol. J. Phys. Chem. A 2015, 119, 10349–10359. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Guzman, M.I. Oxidation of substituted catechols at the air–water interface: Production of carboxylic acids, quinones, and polyphenols. Environ. Sci. Technol. 2017, 51, 4951–4959. [Google Scholar] [CrossRef] [PubMed]
- Rudich, Y.; Donahue, N.M.; Mentel, T.F. Aging of organic aerosol: Bridging the gap between laboratory and field studies. Ann. Rev. Phys. Chem. 2007, 58, 321–352. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Updyke, K.M.; Nguyen, T.B.; Nizkorodov, S.A. Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors. Atmos. Environ. 2012, 63, 22–31. [Google Scholar] [CrossRef]
- Cappa, C.D.; Che, D.L.; Kessler, S.H.; Kroll, J.H.; Wilson, K.R. Variations in organic aerosol optical and hygroscopic properties upon heterogeneous OH oxidation. J. Geophys. Res. Atmos. 2011, 116. [Google Scholar] [CrossRef] [Green Version]
- Bond, T.C. Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion. Geophys. Res. Lett. 2001, 28, 4075–4078. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.E.; Ramanathan, V.; Decremer, D. Observationally constrained estimates of carbonaceous aerosol radiative forcing. Proc. Natl. Acad. Sci. USA 2012, 109, 11624–11629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, I.J.; Abbatt, J.P.D. Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat. Chem. 2010, 2, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Kolb, C.E.; Cox, R.A.; Abbatt, J.P.D.; Ammann, M.; Davis, E.J.; Donaldson, D.J.; Garrett, B.C.; George, C.; Griffiths, P.T.; Hanson, D.R.; et al. An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds. Atmos. Chem. Phys. 2010, 10, 10561–10605. [Google Scholar] [CrossRef] [Green Version]
- Pöschl, U.; Rudich, Y.; Ammann, M. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—Part 1: General equations, parameters, and terminology. Atmos. Chem. Phys. 2007, 7, 5989–6023. [Google Scholar] [CrossRef]
- Rader, D.J.; Grasser, T.W.; Castaneda, J.N.; Trott, W.M.; Torczynski, J.R. Measurements of Thermal Accommodation Coefficients; Department of Energy: Livermore, CA, USA, 2005. [Google Scholar]
- Davidovits, P.; Worsnop, D.R.; Jayne, J.T.; Kolb, C.E.; Winkler, P.; Vrtala, A.; Wagner, P.E.; Kulmala, M.; Lehtinen, K.E.J.; Vesala, T.; et al. Mass accommodation coefficient of water vapor on liquid water. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef] [Green Version]
- Vieceli, J.; Roeselová, M.; Potter, N.; Dang, L.X.; Garrett, B.C.; Tobias, D.J. Molecular dynamics simulations of atmospheric oxidants at the air−water interface: Solvation and accommodation of OH and O3. J. Phys. Chem. B 2005, 109, 15876–15892. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.N.; Ammann, M.; Cox, R.A.; Hynes, R.G.; Jenkin, M.E.; Mellouki, A.; Rossi, M.J.; Troe, J.; Wallington, T.J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V—Heterogeneous reactions on solid substrates. Atmos. Chem. Phys. 2010, 10, 9059–9223. [Google Scholar] [CrossRef] [Green Version]
- Najera, J.J.; Percival, C.J.; Horn, A.B. Infrared spectroscopic studies of the heterogeneous reaction of ozone with dry maleic and fumaric acid aerosol particles. Phys. Chem. Chem. Phys. 2009, 11, 9093–9103. [Google Scholar] [CrossRef] [PubMed]
- Davidovits, P.; Kolb, C.E.; Williams, L.R.; Jayne, J.T.; Worsnop, D.R. Update 1 of: Mass Accommodation and Chemical Reactions at Gas−Liquid Interfaces. Chem. Rev. 2011, 111. [Google Scholar] [CrossRef]
- Ammann, M.; Pöschl, U. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—Part 2: Exemplary practical applications and numerical simulations. Atmos. Chem. Phys. 2007, 7, 6025–6045. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Tachibana, E.; Okuzawa, K.; Aggarwal, S.G.; Kanaya, Y.; Wang, Z.F. High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountaintop aerosols over the North China Plain during wheat burning season. Atmos. Chem. Phys. 2013, 13, 8285–8302. [Google Scholar] [CrossRef] [Green Version]
- Mkoma, S.L.; Kawamura, K. Molecular composition of dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls and fatty acids in atmospheric aerosols from Tanzania, East Africa during wet and dry seasons. Atmos. Chem. Phys. 2013, 13, 2235–2251. [Google Scholar] [CrossRef] [Green Version]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Aqueous photochemistry of glyoxylic acid. J. Phys. Chem. A 2016, 120, 3817–3826. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Guzman, M.I. Reactivity of ketyl and acetyl radicals from direct solar actinic photolysis of aqueous pyruvic acid. J. Phys. Chem. A 2017, 121, 2924–2935. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Arey, J. Atmospheric degradation of volatile organic compounds. Chem. Rev. 2003, 103, 4605–4638. [Google Scholar] [CrossRef] [PubMed]
- Pankow, J.F.; Asher, W.E. SIMPOL.1: A simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. Discuss. 2007, 7, 11839–11894. [Google Scholar] [CrossRef]
- Barsanti, K.C.; Pankow, J.F. Thermodynamics of the formation of atmospheric organic particulate matter by accretion reactions—Part 1: Aldehydes and ketones. Atmos. Environ. 2004, 38, 4371–4382. [Google Scholar] [CrossRef]
- Li, Y.J.; Cheong, G.Y.L.; Lau, A.P.S.; Chan, C.K. Acid-catalyzed condensed-phase reactions of limonene and terpineol and their impacts on gas-to-particle partitioning in the formation of organic aerosols. Environ. Sci. Technol. 2010, 44, 5483–5489. [Google Scholar] [CrossRef] [PubMed]
- Liggio, J.; Li, S.-M.; Brook, J.R.; Mihele, C. Direct polymerization of isoprene and a-pinene on acidic aerosols. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Boyd, C.M.; Sanchez, J.; Xu, L.; Eugene, A.J.; Nah, T.; Tuet, W.Y.; Guzman, M.I.; Ng, N.L. Secondary organic aerosol formation from the β-pinene+NO3 system: Effect of humidity and peroxy radical fate. Atmos. Chem. Phys. 2015, 15, 7497–7522. [Google Scholar] [CrossRef]
- Williams, M.B.; Michelsen, R.R.H.; Axson, J.L.; Iraci, L.T. Uptake of acetone, acetaldehyde and ethanol in cold sulfuric acid solutions containing organic material: Carbon accretion mechanisms. Atmos. Environ. 2010, 44, 1145–1151. [Google Scholar] [CrossRef]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Optical absorptivity versus molecular composition of model organic aerosol matter. J. Phys. Chem. A 2009, 113, 10512–10520. [Google Scholar] [CrossRef] [PubMed]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Thermochromism of model organic aerosol matter. J. Phys. Chem. Lett. 2010, 1, 368–373. [Google Scholar] [CrossRef]
- Guzmán, M.I.; Colussi, A.J.; Hoffmann, M.R. Photoinduced oligomerization of aqueous pyruvic acid. J. Phys. Chem. A 2006, 110, 3619–3626. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Guzman, M.I. Reply to “Comment on ‘Reactivity of ketyl and acetyl radicals from direct solar actinic photolysis of aqueous pyruvic acid’”. J. Phys. Chem. A 2017, 121, 8741–8744. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Pillar, E.A.; Colussi, A.J.; Guzman, M.I. Enhanced acidity of acetic and pyruvic acids on the surface of water. Langmuir 2018, 34, 9307–9713. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Negative production of acetoin in the photochemistry of aqueous pyruvic acid. Proc. Natl. Acad. Sci. USA 2013, 110, E4274–E4275. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.-S.; Eugene, A.J.; Guzman, M.I. Cross photoreaction of glyoxylic and pyruvic acids in model aqueous aerosol. J. Phys. Chem. A 2018, 122, 6457–6466. [Google Scholar] [CrossRef] [PubMed]
- Canonica, S.; Kohn, T.; Mac, M.; Real, F.J.; Wirz, J.; von Gunten, U. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ. Sci. Technol. 2005, 39, 9182–9188. [Google Scholar] [CrossRef] [PubMed]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E.; Harrison, M.A.J.; Olariu, R.-I.; Arsene, C. Photochemical reactions in the tropospheric aqueous phase and on particulate matter. Chem. Soc. Rev. 2006, 35, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Colussi, A.; Hoffmann, M.R. Photogeneration of distant radical pairs in aqueous pyruvic acid glasses. J. Phys. Chem. A 2006, 110, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Hoffmann, M.R.; Colussi, A.J. Photolysis of pyruvic acid in ice: Possible relevance to CO and CO2 ice core record anomalies. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.J.; MacDonald, S.M.; Shaw, M.D.; Kumar, R.; Saunders, R.W.; Parthipan, R.; Wilson, J.; Plane, J.M.C. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 2013, 6, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Kroll, J.H.; Smith, J.D.; Che, D.L.; Kessler, S.H.; Worsnop, D.R.; Wilson, K.R. Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol. Phys. Chem. Chem. Phys. 2009, 11, 8005–8014. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; He, K.; Duan, F.; Cheng, Y.; Ma, Y. Measurement of humic-like substances in aerosols: A review. Environ. Pollut. 2013, 181, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, C.M.; Aeschbacher, M.; Page, S.E.; Wenk, J.; Sander, M.; McNeill, K. Photooxidation-induced changes in optical, electrochemical, and photochemical properties of humic substances. Environ. Sci. Technol. 2014, 48, 2688–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillar, E.A.; Guzman, M.I.; Rodriguez, J.M. Conversion of iodide to hypoiodous acid and iodine in aqueous microdroplets exposed to ozone. Environ. Sci. Technol. 2013, 47, 10971–10979. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.E. A constant-angle mie scattering method (CAMS) for investigation of particle formation processes. J. Colloid Interface Sci. 1985, 105, 456–467. [Google Scholar] [CrossRef]
- Kupc, A.; Winkler, P.M.; Vrtala, A.; Wagner, P.E. Unusual temperature dependence of heterogeneous nucleation of water vapor on ag particles. Aerosol. Sci. Technol. 2013, 47, I–IV. [Google Scholar] [CrossRef]
- Vesala, T.; Kulmala, M.; Rudolf, R.; Vrtala, A.; Wagner, P.E. Models for condensational growth and evaporation of binary aerosol particles. J. Aerosol. Sci. 1997, 28, 565–598. [Google Scholar] [CrossRef]
- Quinlan, M.A.; Reihs, C.M.; Golden, D.M.; Tolbert, M.A. Heterogeneous reactions on model polar stratospheric cloud surfaces: Reaction of dinitrogen pentoxide on ice and nitric acid trihydrate. J. Phys. Chem. 1990, 94, 3255–3260. [Google Scholar] [CrossRef]
- Caloz, F.; Fenter, F.F.; Tabor, K.D.; Rossi, M.J. Paper I: Design and construction of a Knudsen-cell reactor for the study of heterogeneous reactions over the temperature range 130–750 K: Performances and limitations. Rev. Sci. Instrum. 1997, 68, 3172–3179. [Google Scholar] [CrossRef]
- Worsnop, D.R.; Zahniser, M.S.; Kolb, C.E.; Gardner, J.A.; Watson, L.R.; Van Doren, J.M.; Jayne, J.T.; Davidovits, P. The temperature dependence of mass accommodation of sulfur dioxide and hydrogen peroxide on aqueous surfaces. J. Phys. Chem. 1989, 93, 1159–1172. [Google Scholar] [CrossRef]
- Li, Y.Q.; Davidovits, P.; Kolb, C.E.; Worsnop, D.R. Mass and thermal accommodation coefficients of H2O (g) on liquid water as a function of temperature. J. Phys. Chem. A 2001, 105, 10627–10634. [Google Scholar] [CrossRef]
- Worsnop, D.R.; Shi, Q.; Jayne, J.T.; Kolb, C.E.; Swartz, E.; Davidovits, P. Gas-phase diffusion in droplet train measurements of uptake coefficients. J. Aerosol. Sci. 2001, 32, 877–891. [Google Scholar] [CrossRef]
- Ryder, O.S.; Campbell, N.R.; Morris, H.; Forestieri, S.; Ruppel, M.J.; Cappa, C.D.; Tivanski, A.; Prather, K.; Bertram, T.H. Role of organic coatings in regulating N2O5 reactive uptake to sea spray aerosol. J. Phys. Chem. A 2015, 119, 11683–11692. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, T.; Abbatt, J.P.D. Heterogeneous reaction of ozone with liquid unsaturated fatty acids: Detailed kinetics and gas-phase product studies. Phys. Chem. Chem. Phys. 2004, 6, 84–93. [Google Scholar] [CrossRef]
- McCabe, J.; Abbatt, J.P.D. Heterogeneous loss of gas-phase ozone on n-hexane soot surfaces: Similar kinetics to loss on other chemically unsaturated solid surfaces. J. Phys. Chem. C 2009, 113, 2120–2127. [Google Scholar] [CrossRef]
- Arangio, A.M.; Slade, J.H.; Berkemeier, T.; Pöschl, U.; Knopf, D.A.; Shiraiwa, M. Multiphase chemical kinetics of OH radical uptake by molecular organic markers of biomass burning aerosols: Humidity and temperature dependence, surface reaction, and bulk diffusion. J. Phys. Chem. A 2015, 119, 4533–4544. [Google Scholar] [CrossRef] [PubMed]
- Garrett, B.C.; Schenter, G.K.; Morita, A. Molecular simulations of the transport of molecules across the liquid/vapor interface of water. Chem. Rev. 2006, 106, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Guascito, M.R.; Cesari, D.; Chirizzi, D.; Genga, A.; Contini, D. XPS surface chemical characterization of atmospheric particles of different sizes. Atmos. Environ. 2015, 116, 146–154. [Google Scholar] [CrossRef]
- Kirchner, U.; Vogt, R.; Natzeck, C.; Goschnick, J. Single particle MS, SNMS, SIMS, XPS, and FTIR spectroscopic analysis of soot particles during the AIDA campaign. J. Aerosol. Sci. 2003, 34, 1323–1346. [Google Scholar] [CrossRef]
- Qi, J.; Feng, L.; Li, X.; Zhang, M. An X-ray photoelectron spectroscopy study of elements on the surface of aerosol particles. J. Aerosol. Sci. 2006, 37, 218–227. [Google Scholar] [CrossRef]
- Faude, F.; Goschnick, J. XPS, SIMS and SNMS applied to a combined analysis of aerosol particles from a region of considerable air pollution in the upper Rhine valley. Fresen. J. Anal. Chem. 1997, 358, 67–72. [Google Scholar] [CrossRef]
- Washenfelder, R.A.; Attwood, A.R.; Brock, C.A.; Guo, H.; Xu, L.; Weber, R.J.; Ng, N.L.; Allen, H.M.; Ayres, B.R.; Baumann, K.; et al. Biomass burning dominates brown carbon absorption in the rural southeastern United States. Geophys. Res. Lett. 2015, 42, 653–664. [Google Scholar] [CrossRef] [Green Version]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.M.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, M.; Axisa, D.; Tompa, É.; Freney, E.; Bruintjes, R.; Buseck, P.R. Interactions of mineral dust with pollution and clouds: An individual-particle TEM study of atmospheric aerosol from Saudi Arabia. Atmos. Res. 2013, 122, 347–361. [Google Scholar] [CrossRef]
- O’Brien, R.E.; Wang, B.; Kelly, S.T.; Lundt, N.; You, Y.; Bertram, A.K.; Leone, S.R.; Laskin, A.; Gilles, M.K. Liquid–liquid phase separation in aerosol particles: Imaging at the nanometer scale. Environ. Sci. Technol. 2015, 49, 4995–5002. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, P.G.; Yadav, S. Characterization of PM2.5 by X-ray diffraction and scanning electron microscopy—Energy dispersive spectrometer: Its relation with different pollution sources. Int. J. Environ. Sci. Technol. 2014, 11, 217–232. [Google Scholar] [CrossRef]
- Sakata, K.; Sakaguchi, A.; Tanimizu, M.; Takaku, Y.; Yokoyama, Y.; Takahashi, Y. Identification of sources of lead in the atmosphere by chemical speciation using X-ray absorption near-edge structure (XANES) spectroscopy. J. Environ. Sci. 2014, 26, 343–352. [Google Scholar] [CrossRef]
- Isaacman-VanWertz, G.; Massoli, P.; O’Brien, R.; Lim, C.; Franklin, J.P.; Moss, J.A.; Hunter, J.F.; Nowak, J.B.; Canagaratna, M.R.; Misztal, P.K.; et al. Chemical evolution of atmospheric organic carbon over multiple generations of oxidation. Nat. Chem. 2018, 10, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Katrib, Y.; Biskos, G.; Buseck, P.R.; Davidovits, P.; Jayne, J.T.; Mochida, M.; Wise, M.E.; Worsnop, D.R.; Martin, S.T. Ozonolysis of mixed oleic-acid/stearic-acid particles: Reaction kinetics and chemical morphology. J. Phys. Chem. A 2005, 109, 10910–10919. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Athalye, R.R.; Rodriguez, J.M. Concentration effects and ion properties controlling the fractionation of halides during aerosol formation. J. Phys. Chem. A 2012, 116, 5428–5435. [Google Scholar] [CrossRef] [PubMed]
- Iribarne, J.V.; Thomson, B.A. On the evaporation of small ions from charged droplets. J. Chem. Phys. 1976, 64, 2287–2294. [Google Scholar] [CrossRef]
- Barnum, T.J.; Medeiros, N.; Hinrichs, R.Z. Condensed-phase versus gas-phase ozonolysis of catechol: A combined experimental and theoretical study. Atmos. Environ. 2012, 55, 98–106. [Google Scholar] [CrossRef]
- von Sonntag, C.; von Gunten, U. Chemistry of Ozone in Water and Wastewater Treatment; IWA Publishing: London, UK, 2012. [Google Scholar]
- Bertram, T.H.; Thornton, J.A.; Riedel, T.P. An experimental technique for the direct measurement of N2O5 reactivity on ambient particles. Atmos. Meas. Technol. 2009, 2, 231–242. [Google Scholar] [CrossRef]
- Morris, J.W.; Davidovits, P.; Jayne, J.T.; Jimenez, J.L.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Barney, W.S.; Cass, G. Kinetics of submicron oleic acid aerosols with ozone: A novel aerosol mass spectrometric technique. Geophys. Res. Lett. 2002, 29, 71–74. [Google Scholar] [CrossRef]
- Sage, A.M.; Weitkamp, E.A.; Robinson, A.L.; Donahue, N.M. Reactivity of oleic acid in organic particles: Changes in oxidant uptake and reaction stoichiometry with particle oxidation. Phys. Chem. Chem. Phys. 2009, 11, 7951–7962. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, P.J. Aerosol products, mechanisms, and kinetics of heterogeneous reactions of ozone with oleic acid in pure and mixed particles. Faraday Discuss. 2005, 130, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.S.; Ziemann, P.J. Reaction of oleic acid particles with NO3 radicals: Products, mechanism, and implications for radical-initiated organic aerosol oxidation. J. Phys. Chem. A 2006, 110, 3567–3577. [Google Scholar] [CrossRef] [PubMed]
- Last, D.J.; Najera, J.J.; Percival, C.J.; Horn, A.B. A comparison of infrared spectroscopic methods for the study of heterogeneous reactions occurring on atmospheric aerosol proxies. Phys. Chem. Chem. Phys. 2009, 11, 8214–8225. [Google Scholar] [CrossRef] [PubMed]
- Leng, C.; Hiltner, J.; Pham, H.; Kelley, J.; Mach, M.; Zhang, Y.; Liu, Y. Kinetics study of heterogeneous reactions of ozone with erucic acid using an ATR-IR flow reactor. Phys. Chem. Chem. Phys. 2014, 16, 4350–4360. [Google Scholar] [CrossRef] [PubMed]
- Sillman, S. The relation between ozone, NOx and hydrocarbons in urban and polluted rural environments. Atmos. Environ. 1999, 33, 1821–1845. [Google Scholar] [CrossRef]
- Atkinson, R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. [Google Scholar] [CrossRef]
- Pankow, J.F. An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol. Atmos. Environ. 1994, 28, 189–193. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Kroll, J.H.; Ng, N.L.; Seinfeld, J.H. Kinetic modeling of secondary organic aerosol formation: Effects of particle- and gas-phase reactions of semivolatile products. Atmos. Chem. Phys. 2007, 7, 4135–4147. [Google Scholar] [CrossRef]
- Andreae, M.O.; Gelencsér, A. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos. Chem. Phys. 2006, 6, 3131–3148. [Google Scholar] [CrossRef] [Green Version]
- Enami, S.; Hoffmann, M.R.; Colussi, A.J. How phenol and α-tocopherol react with ambient ozone at gas/liquid interfaces. J. Phys. Chem. A 2009, 113, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Enami, S.; Vecitis, C.D.; Cheng, J.; Hoffmann, M.R.; Colussi, A.J. Global inorganic source of atmospheric bromine. J. Phys. Chem. A 2007, 111, 8749–8752. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhang, J.; Cai, S.; Liao, Y.; Zhao, W.; Hu, C.; Gu, X.; Fang, L.; Zhang, W. Mass spectrometric study of aged benzene secondary organic aerosol in the presence of dry ammonium sulfate. J. Atmos. Chem. 2016, 73, 329–344. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. Introductory lecture: Atmospheric chemistry in the Anthropocene. Faraday Discuss. 2017, 200, 11–58. [Google Scholar] [CrossRef] [PubMed]
- Lavi, A.; Lin, P.; Bhaduri, B.; Carmieli, R.; Laskin, A.; Rudich, Y. Characterization of light-absorbing oligomers from reactions of phenolic compounds and Fe(III). ACS Earth Space Chem. 2017, 1, 637–646. [Google Scholar] [CrossRef]
- Sun, J.; Mei, Q.; Wei, B.; Huan, L.; Xie, J.; He, M. Mechanisms for ozone-initiated removal of biomass burning products from the atmosphere. Environ. Chem. 2018, 15, 83–91. [Google Scholar] [CrossRef]
- Magalhães, A.C.O.; Esteves da Silva, J.C.G.; Pinto da Silva, L. Density functional theory calculation of the absorption properties of brown carbon chromophores generated by catechol heterogeneous ozonolysis. ACS Earth Space Chem. 2017, 1, 353–360. [Google Scholar] [CrossRef]
- Sarwar, G.; Gantt, B.; Schwede, D.; Foley, K.; Mathur, R.; Saiz-Lopez, A. Impact of enhanced ozone deposition and halogen chemistry on tropospheric ozone over the northern hemisphere. Environ. Sci. Technol. 2015, 49, 9203–9211. [Google Scholar] [CrossRef] [PubMed]
- Holla, R.; Schmitt, S.; Frieß, U.; Pöhler, D.; Zingler, J.; Corsmeier, U.; Platt, U. Vertical distribution of BrO in the boundary layer at the Dead Sea. Environ. Chem. 2015, 12, 438–460. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Li, Z.; Zhang, Y.; Ge, M. Gas-phase hydration of glyoxylic acid: Kinetics and atmospheric implications. Chemosphere 2017, 186, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, E.G.; Abbatt, J.P.D. Heterogeneous OH oxidation of secondary brown carbon aerosol. Atmos. Chem. Phys. Discuss. 2018, 2018, 1–29. [Google Scholar] [CrossRef]
- Saunders, R.W.; Kumar, R.; MacDonald, S.M.; Plane, J.M.C. Insights into the photochemical transformation of iodine in aqueous systems: Humic acid photosensitized reduction of iodate. Environ. Sci. Technol. 2012, 46, 11854–11861. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.D.; Estillore, A.D.; Morris, H.S.; Ray, K.K.; Alejandro, A.; Grassian, V.H.; Tivanski, A.V. Direct surface tension measurements of individual sub-micrometer particles using atomic force microscopy. J. Phys. Chem. A 2017, 121, 8296–8305. [Google Scholar] [CrossRef] [PubMed]
- Nissenson, P.; Wingen, L.M.; Hunt, S.W.; Finlayson-Pitts, B.J.; Dabdub, D. Rapid formation of molecular bromine from deliquesced NaBr aerosol in the presence of ozone and UV light. Atmos. Environ. 2014, 89, 491–506. [Google Scholar] [CrossRef]
- Desyaterik, Y.; Sun, Y.; Shen, X.; Lee, T.; Wang, X.; Wang, T.; Collett, J.L. Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China. J. Geophys. Res. Atmos. 2013, 118, 7389–7399. [Google Scholar] [CrossRef] [Green Version]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Hoque, M.; Kawamura, K.; Seki, O.; Hoshi, N. Spatial distributions of dicarboxylic acids, ω-oxoacids, pyruvic acid and α-dicarbonyls in the remote marine aerosols over the North Pacific. Mar. Chem. 2015, 172, 1–11. [Google Scholar] [CrossRef]
- Veres, P.; Roberts, J.M.; Burling, I.R.; Warneke, C.; de Gouw, J.; Yokelson, R.J. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry. J. Geophys. Res. Atmos. 2010, 115. [Google Scholar] [CrossRef] [Green Version]
- Henze, D.K.; Seinfeld, J.H.; Ng, N.L.; Kroll, J.H.; Fu, T.M.; Jacob, D.J.; Heald, C.L. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: High- vs. low-yield pathways. Atmos. Chem. Phys. 2008, 8, 2405–2420. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Engling, G.; Yu, J.Z. Humic-like substances in fresh emissions of rice straw burning and in ambient aerosols in the Pearl River Delta Region, China. Atmos. Chem. Phys. 2010, 10, 6487–6500. [Google Scholar] [CrossRef]
- Hoffer, A.; Gelencsér, A.; Guyon, P.; Kiss, G.; Schmid, O.; Frank, G.P.; Artaxo, P.; Andreae, M.O. Optical properties of humic-like substances (HULIS) in biomass-burning aerosols. Atmos. Chem. Phys. 2006, 6, 3563–3570. [Google Scholar] [CrossRef] [Green Version]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillar-Little, E.A.; Guzman, M.I. An Overview of Dynamic Heterogeneous Oxidations in the Troposphere. Environments 2018, 5, 104. https://doi.org/10.3390/environments5090104
Pillar-Little EA, Guzman MI. An Overview of Dynamic Heterogeneous Oxidations in the Troposphere. Environments. 2018; 5(9):104. https://doi.org/10.3390/environments5090104
Chicago/Turabian StylePillar-Little, Elizabeth A., and Marcelo I. Guzman. 2018. "An Overview of Dynamic Heterogeneous Oxidations in the Troposphere" Environments 5, no. 9: 104. https://doi.org/10.3390/environments5090104
APA StylePillar-Little, E. A., & Guzman, M. I. (2018). An Overview of Dynamic Heterogeneous Oxidations in the Troposphere. Environments, 5(9), 104. https://doi.org/10.3390/environments5090104