
Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Exposure, Absorption and Metabolism
3. Potential Mechanisms of Action
4. Implications of Bisphenol Exposure on the Development of Type 2 Diabetes
5. The Effects of Bisphenols on Skeletal Muscle Glucose Metabolism
5.1. Insulin Signaling Pathway
5.2. Mitochondrial Function
6. The Effects of Bisphenols on Adipose Tissue Metabolism
6.1. Adipogenesis
6.2. Insulin Signaling
6.3. Body Weight, Adipocyte Size, Lipid Accumulation
6.4. Adipokine Signaling
6.5. Adipose Depot Effects
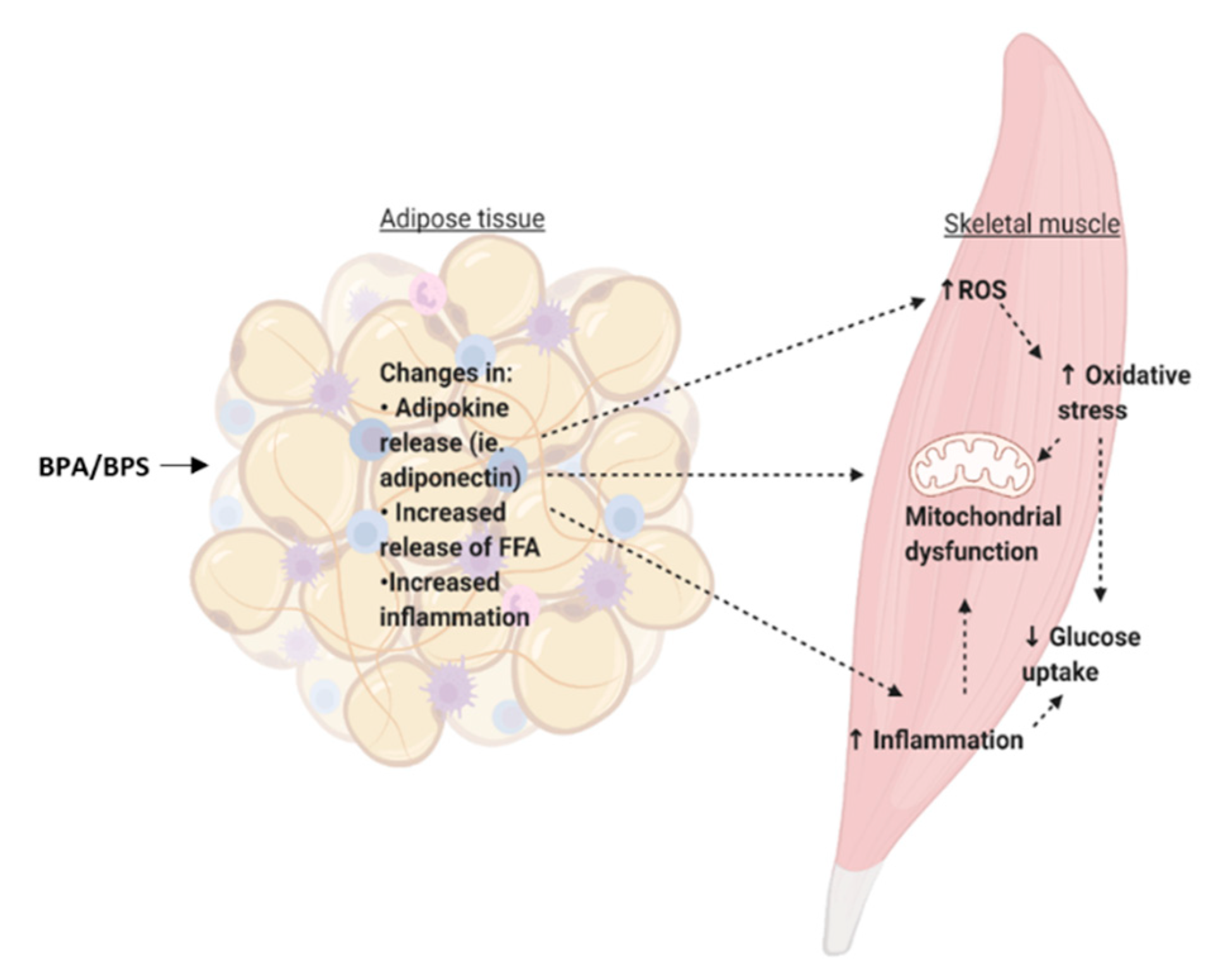
7. Can Bisphenols Alter on the Cross-Talk between the Adipose Tissue and the Skeletal Muscle?
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roglic, G. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Soundararajan, A.; Prabu, P.; Mohan, V.; Gibert, Y.; Balasubramanyam, M. Novel insights of elevated systemic levels of bisphenol-A (BPA) linked to poor glycemic control, accelerated cellular senescence and insulin resistance in patients with type 2 diabetes. Mol. Cell. Biochem. 2019, 458, 171–183. [Google Scholar] [CrossRef]
- Stahlhut, R.W.; Myers, J.P.; Taylor, J.A.; Nadal, A.; Dyer, J.A.; vom Saal, F.S. Experimental BPA exposure and glucose-stimulated insulin response in adult men and women. J. Endocr. Soc. 2018, 2, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Menale, C.; Grandone, A.; Nicolucci, C.; Cirillo, G.; Crispi, S.; di Sessa, A.; Marzuillo, P.; Rossi, S.; Mita, D.G.; Perrone, L.; et al. Bisphenol A is associated with insulin resistance and modulates adiponectin and resistin gene expression in obese children. Pediatric Obes. 2017, 12, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Is the Diabetes Epidemic Primarily Due to Toxins? Integr. Med. 2016, 15, 8–17. [Google Scholar]
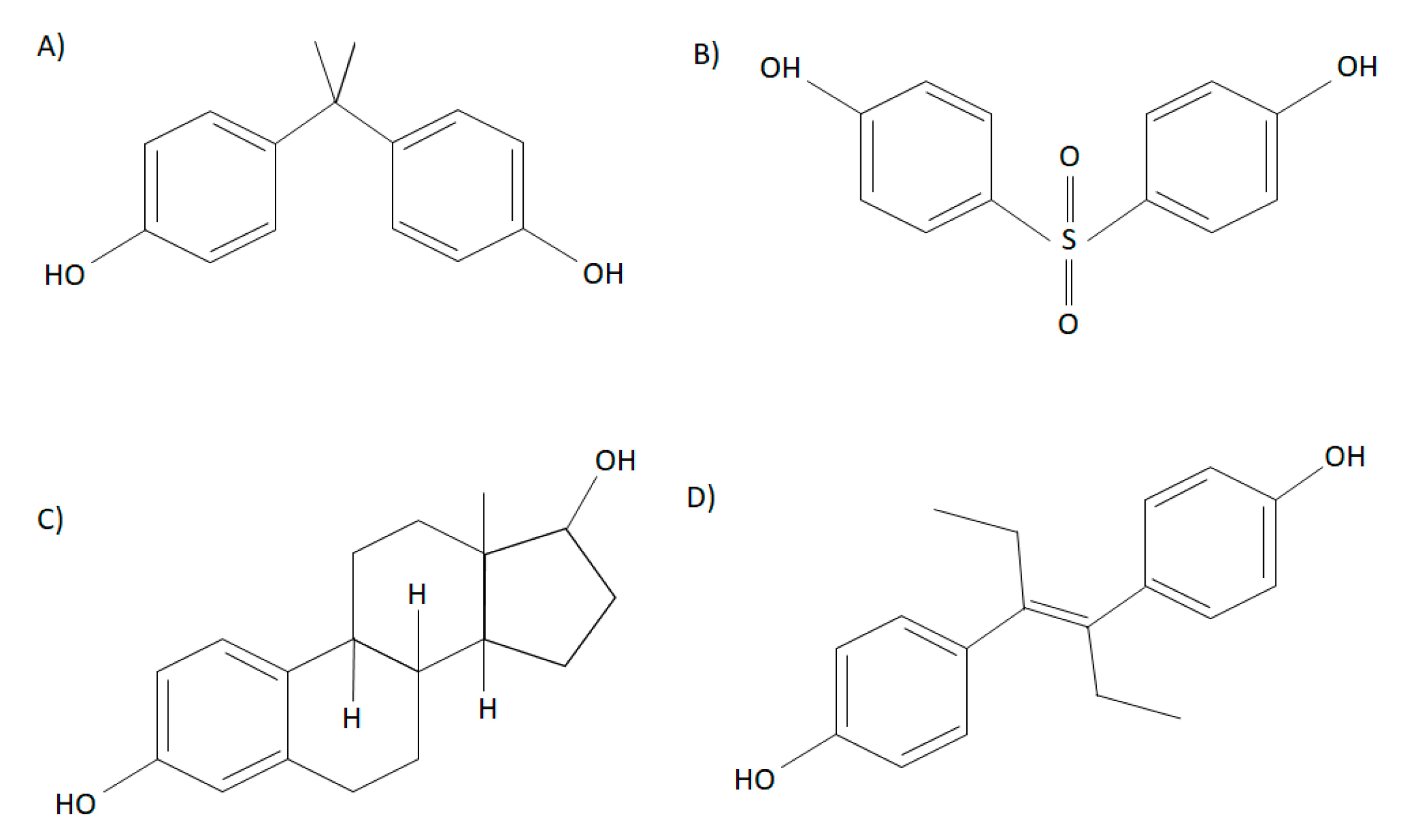
- Ben-Jonathan, N.; Hugo, E.R. Bisphenols come in different flavors: Is “S” better than “A”? Endocrinology 2016, 157, 1321–1323. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.E.; Kendig, E.L.; Belcher, S.M. Assessment of bisphenol A released from reusable plastic, aluminium and stainless steel water bottles. Chemosphere 2011, 85, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geens, T.; Goeyens, L.; Covaci, A. Are potential sources for human exposure to bisphenol-A overlooked? Int. J. Hyg. Environ. Health 2011, 214, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Gould, J.C.; Leonard, L.S.; Maness, S.C.; Wagner, B.L.; Conner, K.; Zacharewski, T.; Safe, S.; McDonnell, D.P.; Gaido, K.W. Bisphenol A interacts with the estrogen receptor h in a distinct manner from estradiol. Mol. Cell. Endocrinol. 1998, 142, 203–214. [Google Scholar] [CrossRef]
- Nakagomi, M.; Suzuki, E.; Saito, Y.; Nagao, T. Endocrine disrupting chemicals, 4-nonylphenol, bisphenol A and butyl benzyl phthalate, impair metabolism of estradiol in male and female rats as assessed by levels of 15α-hydroxyestrogens and catechol estrogens in urine. J. Appl. Toxicol. 2018, 38, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Sajiki, J.; Yonekubo, J. Leaching of bisphenol A (BPA) from polycarbonate plastic to water containing amino acids and its degradation by radical oxygen species. Chemosphere 2004, 55, 861–867. [Google Scholar] [CrossRef]
- Vogel, S.A. The Politics of Plastics: The Making and Unmaking of Bisphenol A “Safety”. Am. J. Public Health 2009, 99, S559–S566. [Google Scholar] [CrossRef]
- Kang, J.S.; Choi, J.S.; Kim, W.K.; Lee, Y.J.; Park, J.W. Estrogenic potency of bisphenol, S. polyethersulfone and their metabolites generated by the rat liver S9 fractions on a MVLN cell using a luciferase reporter gene assay. Reprod. Biol. Endocrinol. 2014, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochester, J.R.; Bolden, A.L. Bisphenol S and F: A systematic review and comparison of the hormonal activity of bisphenol a substitutes. Environ. Health Perspect. 2015, 123, 643–650. [Google Scholar] [CrossRef]
- Ahmed, S.; Atlas, E. Bisphenol S- and bisphenol A-induced adipogenesis of murine preadipocytes occurs through direct peroxisome proliferator-activated receptor gamma activation. Int. J. Obes. 2016, 40, 1566–1573. [Google Scholar] [CrossRef]
- Liao, C.; Kannan, K. Concentrations and profiles of bisphenol a and other bisphenol analogues in foodstuffs from the united states and their implications for human exposure. J. Agric. Food Chem. 2013, 61, 4655–4662. [Google Scholar] [CrossRef]
- Caballero-Casero, N.; Lunar, L.; Rubio, S. Analytical methods for the determination of mixtures of bisphenols and derivatives in human and environmental exposure sources and biological fluids. A review. Anal. Chim. Acta 2016, 908, 22–53. [Google Scholar] [CrossRef]
- Chen, D.; Kannan, K.; Tan, H.; Zheng, Z.; Feng, Y.L.; Wu, Y.; Widelka, M. Bisphenol Analogues Other Than BPA: Environmental Occurrence, Human Exposure, and Toxicity—A Review. Environ. Sci. Technol. 2016, 50, 5438–5453. [Google Scholar] [CrossRef] [PubMed]
- Eladak, S.; Grisin, T.; Moison, D.; Guerquin, M.J.; N’Tumba-Byn, T.; Pozzi-Gaudin, S.; Benachi, A.; Livera, G.; Rouiller-Fabre, V.; Habert, R. A new chapter in the bisphenol a story: Bisphenol S and bisphenol F are not safe alternatives to this compound. Fertil. Steril. 2015, 103, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, L.N.; Chahoud, I.; Heindel, J.J.; Padmanabhan, V.; Paumgartten, F.J.R.; Schoenfelder, G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ. Health Perspect. 2010, 118, 1055–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Department of Health and Human Services. Fourth National Report on Human Exposure to Environmental Chemicals. Updated Tables, January 2019, Volume One, pp. 1–529. Available online: https://www.cdc.gov/exposurereport/pdf/FourthReport_UpdatedTables_Volume1_Jan2019-508.pdf (accessed on 19 April 2021).
- Welshons, W.v.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, 56–69. [Google Scholar] [CrossRef]
- Manukyan, L.; Dunder, L.; Lind, P.M.; Bergsten, P.; Lejonklou, M.H. Developmental exposure to a very low dose of bisphenol A induces persistent islet insulin hypersecretion in Fischer 344 rat offspring. Environ. Res. 2019, 172, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Timms, B.G.; Howdeshell, K.L.; Barton, L.; Bradley, S.; Richter, C.A.; vom Saal, F.S. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc. Natl. Acad. Sci. USA 2005, 102, 7014–7019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, C.; Segatto, M.; Gagliardi, S.; Bertoli, S.; Leone, A.; Barberio, L.; Mandalà, M.; Pallottini, V. Maternal dietary exposure to low-dose bisphenol a affects metabolic and signaling pathways in the brain of rat fetuses. Nutrients 2020, 12, 1448. [Google Scholar] [CrossRef] [PubMed]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Furuta, I.; Kato, E.H.; Kataoka, S.; Usuki, Y.; Kobashi, G.; Sata, F.; Kishi, R.; Fujimoto, S. Maternal serum and amniotic fluid bisphenol A concentrations in the early second trimester. Reprod. Toxicol. 2002, 16, 735–739. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tsutsumi, O.; Ikezuki, Y.; Takai, Y.; Taketani, Y. Positive Relationship between Androgen and the Endocrine Disruptor, Bisphenol A, in Normal Women and Women with Ovarian Dysfunction. Endocr. J. 2004, 51, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Le, H.H.; Carlson, E.M.; Chua, J.P.; Belcher, S.M. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol. Lett. 2008, 176, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Almeida, S.; Raposo, A.; Almeida-González, M.; Carrascosa, C. Bisphenol A: Food Exposure and Impact on Human Health. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1503–1517. [Google Scholar] [CrossRef] [Green Version]
- Bushnik, P.T.; Haines, D.; Levallois, P.; Levesque, J.; Van Oostdom, C.; Viau, C. Lead and bisphenol A concentrations in the Canadian population. Health Rep. 2009, 54, 1547–1554. [Google Scholar]
- VandeVoort, C.A.; Gerona, R.R.; vom Saal, F.S.; Tarantal, A.F.; Hunt, P.A.; Hillenweck, A.; Zalko, D. Maternal and fetal pharmacokinetics of oral radiolabeled and authentic bisphenol a in the rhesus monkey. PLoS ONE 2016, 11, e0165410. [Google Scholar] [CrossRef] [Green Version]
- Schönfelder, G.; Wittfoht, W.; Hopp, H.; Talsness, C.E.; Paul, M.; Chahoud, I. Parent bisphenol a accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 2002, 110, 703–707. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Lemmen, J.G.; Carlsson, B.O.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
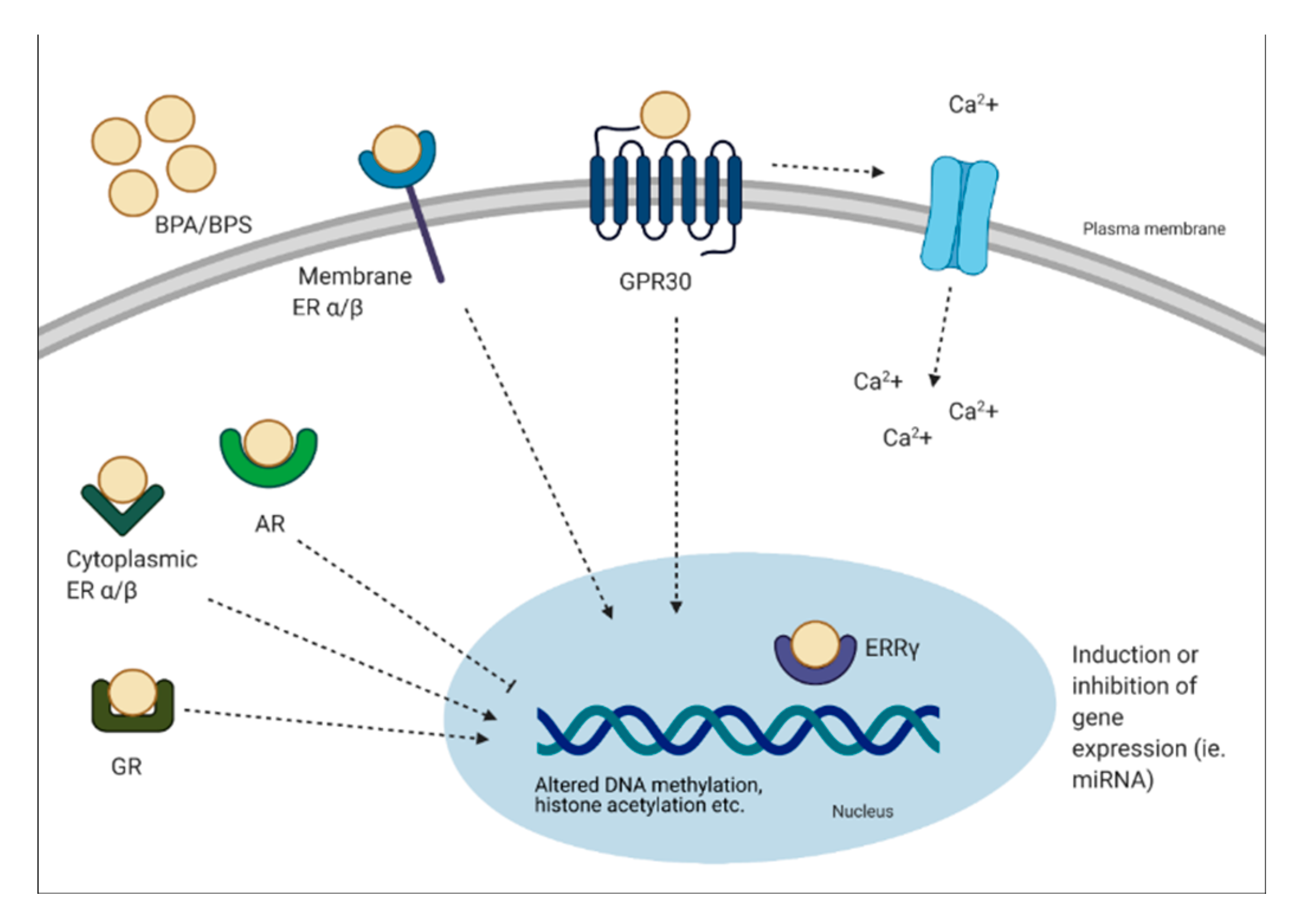
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular mechanisms of action of BPA. Dose-Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heard, D.J.; Norby, P.L.; Holloway, J.; Vissing, H. Human ERRgamm, a Third Member of the Estrogen Receptor-Related Receptor (ERR) Subfamily of Orphan Nuclear Receptors: Tissue-Specific Isoforms Are Expressed during Development and in the Adult. Mol. Endocrinol. 2000, 14, 382–392. [Google Scholar] [PubMed] [Green Version]
- Freitas, J.; Cano, P.; Craig-Veit, C.; Goodson, M.L.; David Furlow, J.; Murk, A.J. Detection of thyroid hormone receptor disruptors by a novel stable in vitro reporter gene assay. Toxicol. Vitr. 2011, 25, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Tagami, T.; Akamizu, T.; Usui, T.; Saijo, M.; Kanamoto, N.; Hataya, Y.; Shimatsu, A.; Kuzuya, H.; Nakao, K. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 2002, 87, 5185–5190. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, Y.J. Bisphenols and thyroid hormone. Endocrinol. Metab. 2019, 34, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Bonefeld-Jørgensen, E.C.; Long, M.; Hofmeister, M.v.; Vinggaard, A.M. Endocrine-disrupting potential of Bisphenol A, Bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: New data and a brief review. Environ. Health Perspect. 2007, 115, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Prasanth, G.K.; Divya, L.M.; Sadasivan, C. Bisphenol-A can bind to human glucocorticoid receptor as an agonist: An in silico study. J. Appl. Toxicol. 2010, 30, 769–774. [Google Scholar] [CrossRef]
- Wozniak, A.L.; Bulayeva, N.N.; Watson, C.S. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ. Health Perspect. 2005, 113, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Alavian-ghavanini, A.; Lin, P.; Lind, P.M.; Rimfors, S.R.; Halin, M.; Dunder, L.; Tang, M.; Lindh, C.; Bornehag, C.-G.; Rüegg, J. Prenatal Bisphenol A Exposure is Linked to Epigenetic Changes in Glutamate Receptor Subunit Gene Grin2b in Female Rats and Humans. Sci. Rep. 2018, 8, 11315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avissar-Whiting, M.; Veiga, K.R.; Uhl, K.M.; Maccani, M.A.; Gagne, L.A.; Moen, E.L.; Marsit, C.J. Bisphenol A exposure leads to specific microRNA alterations in placental cells. Reprod. Toxicol. (Elmsford N. Y.) 2010, 29, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Thakur, M.K. Effect of perinatal exposure to Bisphenol-A on DNA methylation and histone acetylation in cerebral cortex and hippocampus of postnatal male mice. J. Toxicol. Sci. 2017, 42, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Zheng, L.D.; Smith, C.; Luo, J.; Robinson, A.; Almeida, F.A.; Wang, Z.; Olumi, A.F.; Liu, D.; Cheng, Z. Estradiol signaling mediates gender difference in visceral adiposity via autophagy article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Arambula, S.E.; Jima, D.; Patisaul, H.B. Prenatal bisphenol A (BPA) exposure alters the transcriptome of the neonate rat amygdala in a sex-specific manner: A CLARITY-BPA consortium study. Neurotoxicology 2018, 65, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Teppala, S. Relationship between urinary bisphenol A levels and diabetes mellitus. J. Clin. Endocrinol. Metab. 2011, 96, 3822–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadkhaniha, R.; Mansouri, M.; Yunesian, M.; Omidfar, K.; Jeddi, M.Z.; Larijani, B.; Mesdaghinia, A.; Rastkari, N. Association of urinary bisphenol a concentration with type-2 diabetes mellitus. J. Environ. Health Sci. Eng. 2014, 12, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Cornelis, M.C.; Townsend, M.K.; Tobias, D.K.; Heather Eliassen, A.; Franke, A.A.; Hauser, R.; Hu, F.B. Association of urinary concentrations of bisphenol A and phthalate metabolites with risk of type 2 diabetes: A prospective investigation in the nurses’ health study (NHS) and NHSII cohorts. Environ. Health Perspect. 2014, 122, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.K.; O’Neill, M.S.; Sowers, M.F.R.; Park, S.K. Urinary Bisphenol a and type-2 diabetes in U.S. Adults: Data from NHANES 2003–2008. PLoS ONE 2011, 6, e26868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaKind, J.S.; Goodman, M.; Naiman, D.Q. Use of NHANES Data to Link Chemical Exposures to Chronic Diseases: A Cautionary Tale. PLoS ONE 2012, 7, e51086. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol a disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Makaji, E.; Raha, S.; Wade, M.G.; Holloway, A.C. Effect of Environmental Contaminants on Beta Cell Function. Int. J. Toxicol. 2011, 30, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Weldingh, N.M.; Jørgensen-Kaur, L.; Becher, R.; Holme, J.A.; Bodin, J.; Nygaard, U.C.; Bølling, A.K. Bisphenol A Is More Potent than Phthalate Metabolites in Reducing Pancreatic β-Cell Function. BioMed Res. Int. 2017, 2017, 4614379. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Romacho, T.; Elsen, M.; Röhrborn, D.; Eckel, J. Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, R.W.A.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. In Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy; Dove Medical Press Ltd.: Macclesfield, UK, 2014; Volume 7, pp. 55–64. [Google Scholar]
- Storgaard, H.; Song, X.M.; Jensen, C.B.; Madsbad, S.; Bjö, M.; Vaag, A.; Zierath, J.R. Insulin Signal Transduction in Skeletal Muscle From Glucose-Intolerant Relatives With Type 2 Diabetes. Diabetes 2001, 50, 2770–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Neschen, S.; Bilz, S.; Sono, S.; Tsirigotis, D.; Reznick, R.M.; Moore, I.; Nagai, Y.; Samuel, V.; Sebastian, D.; et al. Muscle-Specific IRS-1 Ser 3 Ala Transgenic Mice Are Skeletal Muscle. Diabetes 2008, 57, 2644–2651. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, C.; Tremblay, F.; Asselin, G.; Jacques, H.; Marette, A. Prevention of Skeletal Muscle Insulin Resistance by Dietary Cod Protein in High Fat-Fed Rats. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E62–E71. [Google Scholar] [CrossRef]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jesse, N. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef]
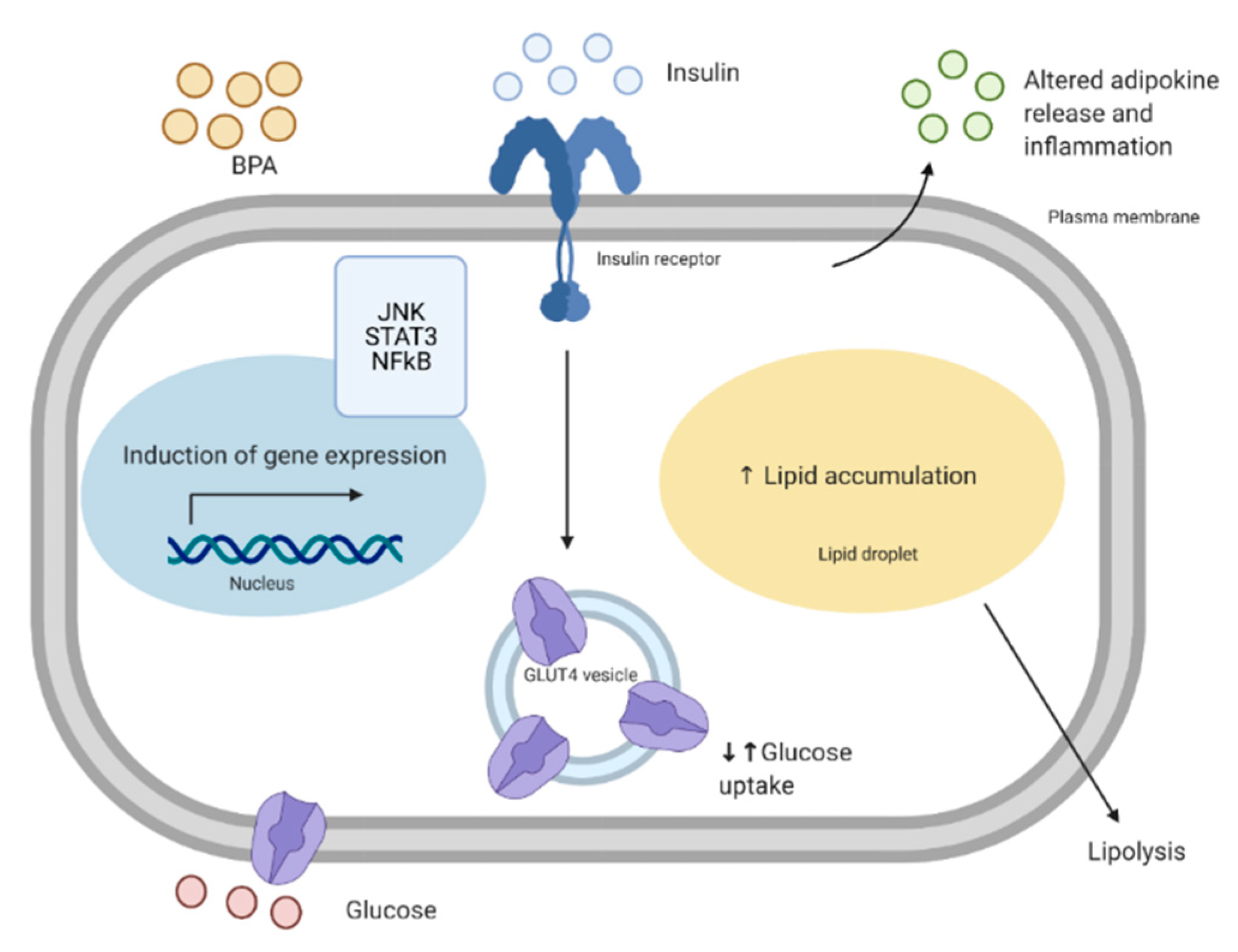
- Mullainadhan, V.; Viswanathan, M.P.; Karundevi, B. Effect of Bisphenol-A (BPA) on insulin signal transduction and GLUT4 translocation in gastrocnemius muscle of adult male albino rat. Int. J. Biochem. Cell Biol. 2017, 90, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Jeong, I.K.; Oh, T.J.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indumathi, D.; Jayashree, S.; Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Akilavalli, N.; Balasubramanian, K. Effect of bisphenol-A on insulin signal transduction and glucose oxidation in skeletal muscle of adult male albino rat. Hum. Exp. Toxicol. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Chehadé, L.; Garneau, L.; Caron, A.; Aguer, C. The effects of acute BPA exposure on skeletal muscle mitochondrial function and glucose metabolism. Mol. Cell. Endocrinol. 2020, 499, 110580. [Google Scholar] [CrossRef]
- Detimary, P.; Gilon, P.; Henquin, J.-C. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: A feedback control mechanism in mouse pancreatic islets. Biochem. J. 1998, 333, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.v.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, E.M.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen-Schimke, J.M.; Guo, Z.K.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; ’t Hart, L.M.; van Essen, E.; Heine, R.J.; Nijpels, G.; Tafrechi, R.S.J.; Raap, A.K.; Janssen, G.M.C.; Lemkes, H.H.P.J. Mitochondrial Diabetes Molecular Mechanisms and Clinical Presentation. Diabetes 2004, 53 (Suppl. 1), S103–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of Acute Changes of Plasma Free Fatty Acids on Intramyocellular Fat Content and Insulin Resistance in Healthy Subjects. Diabetes 2001, 50, 1612–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Qu, W.; Wang, K.; Chen, S.; Zhang, L.; Wu, D.; Chen, Z. Bisphenol A inhibits mucin 2 secretion in intestinal goblet cells through mitochondrial dysfunction and oxidative stress. Biomed. Pharmacother. 2019, 111, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.C.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Aguer, C.; Pasqua, M.; Thrush, A.B.; Moffat, C.; McBurney, M.; Jardine, K.; Zhang, R.; Beauchamp, B.; Dent, R.; McPherson, R.; et al. Increased proton leak and SOD2 expression in myotubes from obese non-diabetic subjects with a family history of type 2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1624–1633. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, X.; Qiu, L.; Wei, J.; Huang, Q.; Fang, C.; Ye, T.; Kang, M.; Shen, H.; Dong, S. Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death Dis. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apovian, C.M.; Okemah, J.; O’Neil, P.M. Body Weight Considerations in the Management of Type 2 Diabetes. Adv. Ther. 2019, 36, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes UK. Diabetes: Facts and Stats. Diabetes UK 2016, 8. Available online: https://mrc.ukri.org/documents/pdf/diabetes-uk-facts-and-stats-june-2015/%0Afile:///C:/Users/sfair/OneDrive/HumanBiology3rdyear/ResearchProject/DissertationWriteUp/DiabetesUK_Facts_Stats_Oct16.pdf (accessed on 20 January 2021).
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol a enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef]
- Boucher, J.G.; Boudreau, A.; Ahmed, S.; Atlas, E. In vitro effects of bisphenol A β-D-glucuronide (BPA-G) on adipogenesis in human and murine preadipocytes. Environ. Health Perspect. 2015, 123, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef]
- Riu, A.; Grimaldi, M.; le Maire, A.; Bey, G.; Phillips, K.; Boulahtouf, A.; Perdu, E.; Zalko, D.; Bourguet, W.; Balaguer, P. Peroxisome proliferator-activated receptor γ is a target for halogenated analogs of bisphenol A. Environ. Health Perspect. 2011, 119, 1227–1232. [Google Scholar] [CrossRef]
- de Filippis, E.; Li, T.; Rosen, E.D. Exposure of adipocytes to bisphenol-A in vitro interferes with insulin action without enhancing adipogenesis. PLoS ONE 2018, 13, e0201122. [Google Scholar] [CrossRef] [Green Version]
- Chamorro-García, R.; Kirchner, S.; Li, X.; Janesick, A.; Casey, S.C.; Chow, C.; Blumberg, B. Bisphenol A diglycidyl ether induces adipogenic differentiation of multipotent stromal stem cells through a peroxisome proliferator-activated receptor gamma-independent mechanism. Environ. Health Perspect. 2012, 120, 984–989. [Google Scholar] [CrossRef]
- Lonnroth, P.; Digirolamo, M.; Krotkiewski, M.; Smith, U. Jnsulin Binding and Responsiveness in Fat Cells from Patients with Reduced Glucose Tolerance and Type II Diabetes. Diabetes 1983, 32, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, A.; Öst, A.; Nystrom, F.H.; Strålfors, P. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J. Biol. Chem. 2005, 280, 34389–34392. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Kawazuma, M.; Adachi, T.; Harigaya, T.; Saito, Y.; Hashimoto, N.; Mori, C. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br. J. Pharmacol. 2004, 141, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Sarsenbayeva, A.; Katsogiannos, P.; Aguer, C.; Pereira, M.J. The effects of bisphenol A and bisphenol S on adipokine expression and glucose metabolism in human adipose tissue. Toxicology 2020, 445, 152600. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. Cyclosporine A and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: Increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J. Clin. Endocrinol. Metab. 2014, 99, E1885–E1894. [Google Scholar] [CrossRef] [Green Version]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.; Ferrini, M.G.; Jellyman, J.K.; Han, G.; Ross, M.G. In vivo and in vitro bisphenol A exposure effects on adiposity. J. Dev. Orig. Health Dis. 2018, 9, 678–687. [Google Scholar] [CrossRef]
- Blesson, C.S.; Yallampalli, C. Pregnancy is a new window of susceptibility for bisphenol a exposure. Endocrinology 2015, 156, 1611–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, Á. Prenatal Exposure to BPA and Offspring Outcomes. Dose-Response 2015, 13, 155932581559039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejonklou, M.H.; Dunder, L.; Bladin, E.; Pettersson, V.; Rönn, M.; Lind, L.; Waldén, T.B.; Lind, P.M. Effects of low-dose developmental bisphenol a exposure on metabolic parameters and gene expression in male and female fischer 344 rat offspring. Environ. Health Perspect. 2017, 125, 067018. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Ahmed, S.; Atlas, E. Bisphenol S induces adipogenesis in primary human preadipocytes from female donors. Endocrinol. 2016, 157, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Héliès-Toussaint, C.; Peyre, L.; Costanzo, C.; Chagnon, M.C.; Rahmani, R. Is bisphenol S a safe substitute for bisphenol A in terms of metabolic function? An in vitro study. Toxicol. Appl. Pharmacol. 2014, 280, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, I.; Oriente, F.; D’esposito, V.; Liguoro, D.; Liguoro, P.; Ambrosio, M.R.; Cabaro, S.; D’Andrea, F.; Beguinot, F.; Formisano, P.; et al. Low-dose bisphenol-a regulates inflammatory cytokines through GPR30 in mammary adipose cells. J. Mol. Endocrinol. 2019, 63, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Hugo, E.R.; Brandebourg, T.D.; Woo, J.G.; Loftus, J.; Alexander, J.W.; Ben-Jonathan, N. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ. Health Perspect. 2008, 116, 1642–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Sopasakis, V.R.; Sandqvist, M.; Gustafson, B.; Hammarstedt, A.; Schmelz, M.; Yang, X.; Jansson, P.-A.; Smith, U. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obes. Res. 2004, 12, 454–460. [Google Scholar] [CrossRef]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) Induces Insulin Resistance in 3T3-L1 Adipocytes and Is, Like IL-8 and Tumor Necrosis Factor-α, Overexpressed in Human Fat Cells from Insulin-resistant Subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and Subcutaneous Adipose Tissues of Obese Subjects Release Interleukin-6: Depot Difference and Regulation by Glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1 mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab. 2014, 307, 289–304. [Google Scholar] [CrossRef]
- Yun Pyo, M.; Ju Kim, H.; Kyung Back, S.; Yang, M. Downregulation of Peritoneal Macrophage Activity in Mice Exposed to Bisphenol A During Pregnancy and Lactation. Arch. Pharm. Res. 2007, 30, 1476–1481. [Google Scholar]
- Lu, X.; Li, M.; Wu, C.; Zhou, C.; Zhang, J.; Zhu, Q.; Shen, T. Bisphenol A promotes macrophage proinflammatory subtype polarization via upregulation of IRF5 expression in vitro. Toxicol. Vitro 2019, 60, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, D.; Krause, K.; Wohland, T.; Gebhardt, C.; Linder, N.; Stumvoll, M.; Blüher, M.; Bechmann, I.; Kovacs, P.; Gericke, M.; et al. Identification of distinct transcriptome signatures of human adipose tissue from fifteen depots. Eur. J. Hum. Genet. 2020, 28, 1714–1725. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Hatazaki, M.; Miyatsuka, T.; Matsuoka, T.; Kajimoto, Y.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Modulation of the JNK Pathway in Liver Affects Insulin Resistance Status. J. Biol. Chem. 2004, 279, 45803–45809. [Google Scholar] [CrossRef] [Green Version]
- Vahdati Hassani, F.; Mehri, S.; Abnous, K.; Birner-Gruenberger, R.; Hosseinzadeh, H. Protective effect of crocin on BPA-induced liver toxicity in rats through inhibition of oxidative stress and downregulation of MAPK and MAPKAP signaling pathway and miRNA-122 expression. Food Chem. Toxicol. 2017, 107, 395–405. [Google Scholar] [CrossRef]
- Lan, H.C.; Lin, I.W.; Yang, Z.J.; Lin, J.H. Low-dose bisphenol A activates Cyp11a1 gene expression and corticosterone secretion in adrenal gland via the JNK signaling pathway. Toxicol. Sci. 2015, 148, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Liu, Y.; Ichikawa, H.; Takemura, S.; Minamiyama, Y. Effects of bisphenol a on oxidative stress in the rat brain. Antioxidants 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Wang, S.; Zhu, W.; Xie, C.; Li, X.; Wu, J.; Zhu, J.; Jiang, Y.; Yang, X.; Li, Y.; et al. Curcumin suppresses JNK pathway to attenuate BPA-induced insulin resistance in LO2 cells. Biomed. Pharmacother. 2018, 97, 1538–1543. [Google Scholar] [CrossRef]
- Wajchenberg, B.L.; Gianella-Neto, D.; da Silva, M.E.R.; Santos, R.F. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm. Metab. Res. 2002, 34, 616–621, undefined. [Google Scholar] [CrossRef]
- Pickering, R.T.; Lee, M.J.; Karastergiou, K.; Gower, A.; Fried, S.K. Depot dependent effects of dexamethasone on gene expression in human omental and abdominal subcutaneous adipose tissues from obese women. PLoS ONE 2016, 11, e0167337. [Google Scholar] [CrossRef]
- Peshdary, V.; Styles, G.; Gagné, R.; Yauk, C.L.; Sorisky, A.; Atlas, E. Depot-Specific Analysis of Human Adipose Cells and Their Responses to Bisphenol S. Endocrinology 2020, 161, bqaa044. [Google Scholar] [CrossRef]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.-K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.-J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin Inhibits Lipolysis in Adipocytes via the Evolutionarily Conserved mTORC1-Egr1-ATGL-Mediated Pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [Green Version]
- Carchia, E.; Porreca, I.; Almeida, P.J.; D’Angelo, F.; Cuomo, D.; Ceccarelli, M.; De Felice, M.; Mallardo, M.; Ambrosino, C. Evaluation of low doses BPA-induced perturbation of glycemia by toxicogenomics points to a primary role of pancreatic islets and to the mechanism of toxicity. Cell Death Disease 2015, 6, e1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almabouada, F.; Diaz-Ruiz, A.; Rabanal-Ruiz, Y.; Peinado, J.R.; Vazquez-Martinez, R.; Malagon, M.M. Adiponectin receptors form homomers and heteromers exhibiting distinct ligand binding and intracellular signaling properties. J. Biol. Chem. 2013, 288, 3112–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 2005, 48, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Planavila, A.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate-induced interleukin 6 production is mediated by protein kinase C and nuclear-factor B activation and leads to glucose transporter 4 down-regulation in skeletal muscle cells. Endocrinology 2005, 146, 3087–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Pathophysiology 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, F.; Pereira, M.J.; Aguer, C. Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue. Environments 2021, 8, 35. https://doi.org/10.3390/environments8040035
Ahmed F, Pereira MJ, Aguer C. Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue. Environments. 2021; 8(4):35. https://doi.org/10.3390/environments8040035
Chicago/Turabian StyleAhmed, Fozia, Maria João Pereira, and Céline Aguer. 2021. "Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue" Environments 8, no. 4: 35. https://doi.org/10.3390/environments8040035
APA StyleAhmed, F., Pereira, M. J., & Aguer, C. (2021). Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue. Environments, 8(4), 35. https://doi.org/10.3390/environments8040035