Reverse Coarsening and the Control of Particle Size Distribution through Surfactant


{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
2. Results and Discussion
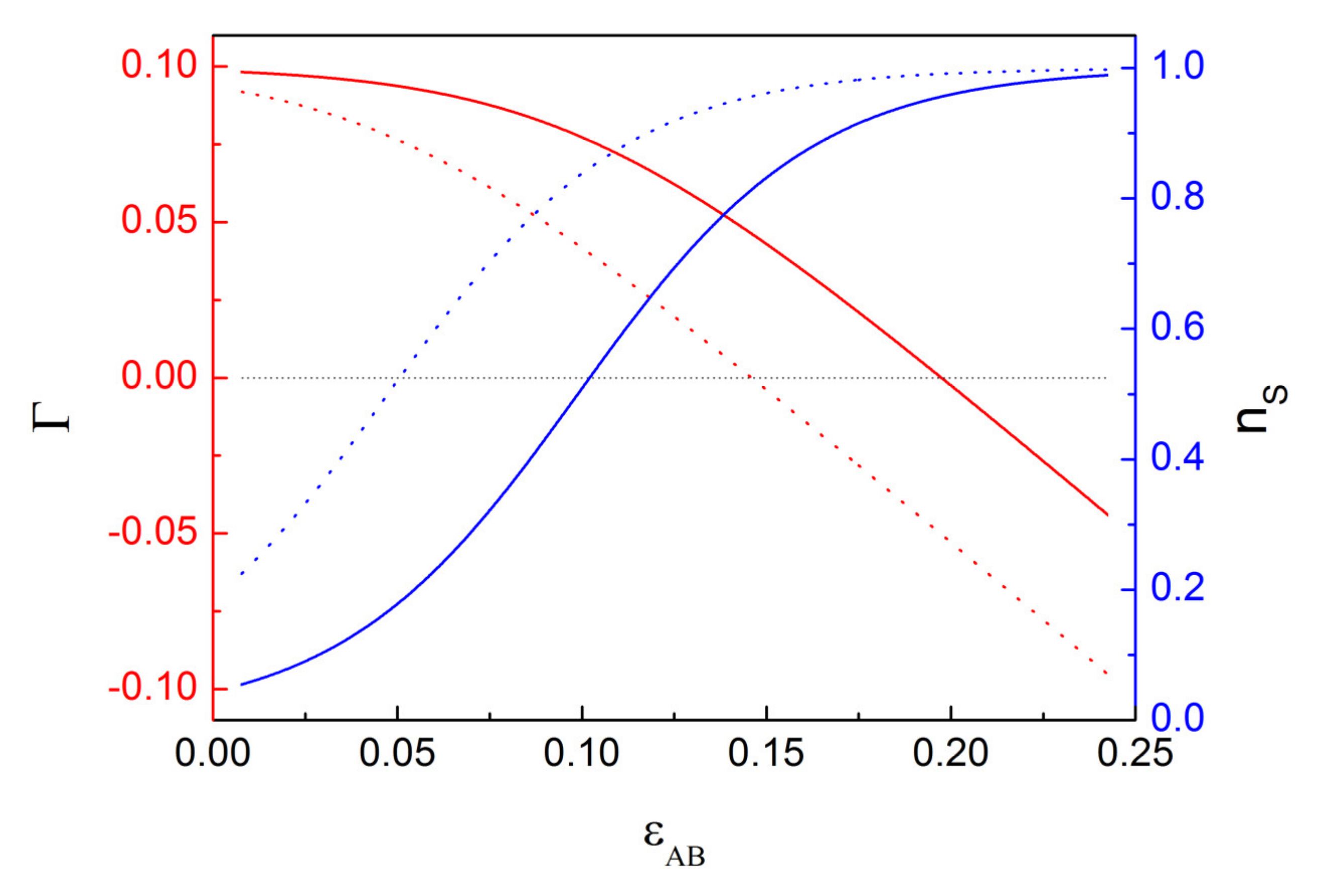
2.1. Isolated Particle in a Binary Solution
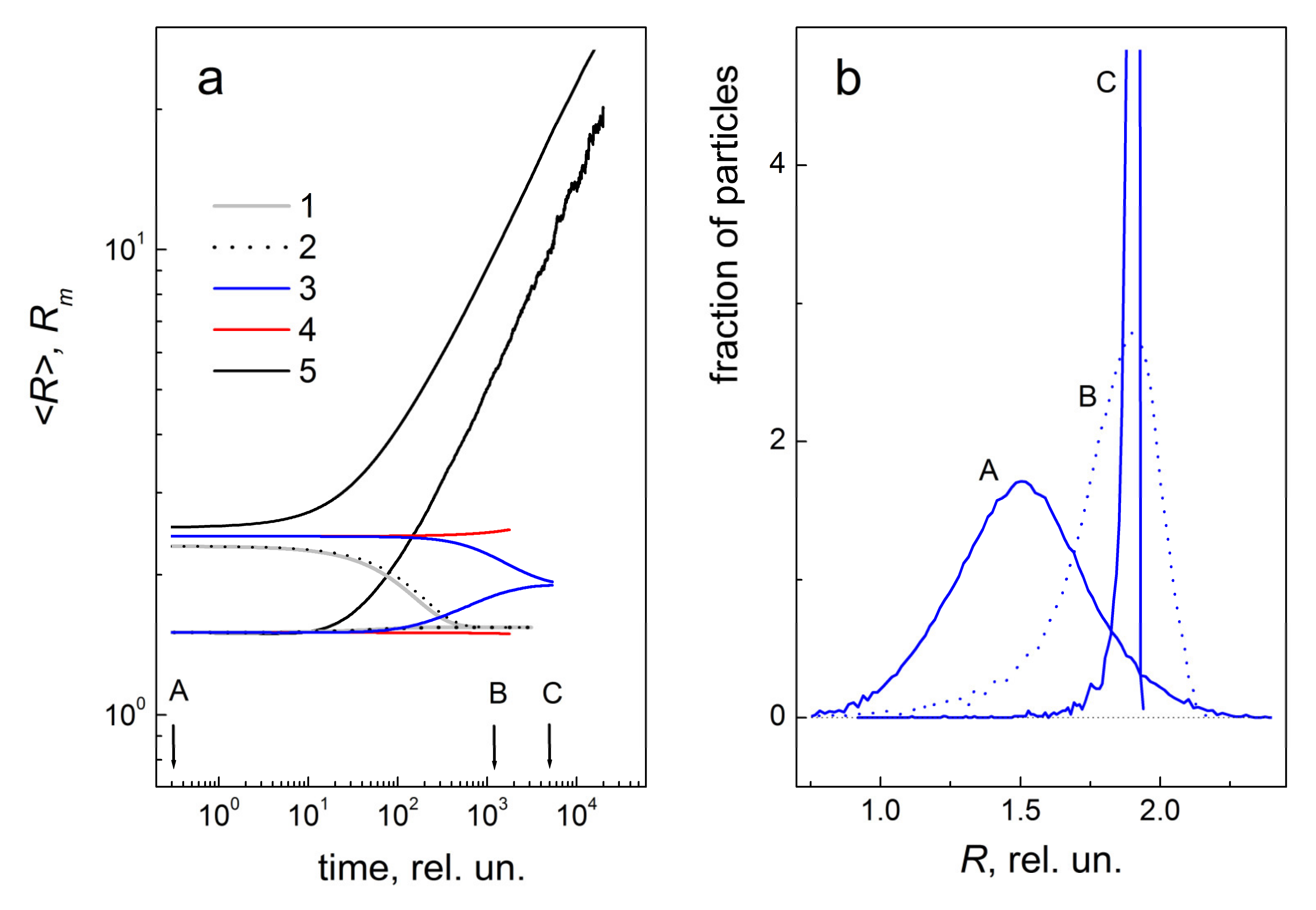
2.2. Simulation of Reverse Coarsening in the Particle Ensemble
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. The Role of Interaction between B-Molecules
Appendix B. Derivation of Main Equations
References
- Chakraverty, B.K. Grain size distribution—1. Conservative systems. J. Phys. Chem. Solids 1967, 28, 2401–2412. [Google Scholar] [CrossRef]
- Prasad, B.L.V.; Stoeva, S.I.; Sorensen, C.M.; Klabunde, K.J. Digestive ripening agents for gold nanoparticles: Alternatives to thiols. Chem. Mater. 2003, 15, 935–942. [Google Scholar] [CrossRef]
- Prasad, B.L.V.; Stoeva, S.I.; Sorensen, C.M.; Klabunde, K.J. Digestive ripening of thiolated gold nanoparticles: The effect of alkyl chain length. Langmuir 2002, 18, 7515–7520. [Google Scholar] [CrossRef]
- Smetana, A.B.; Klabunde, K.J.; Sorensen, C.M. Synthesis of spherical silver nanoparticles by digestive ripening, stabilization with various agents, and their 3-D and 2-D superlattice formation. J. Colloid Interface Sci. 2005, 284, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; El-Ballouli, A.O.; Rollny, L.; Voznyy, O.; Burlakov, V.M.; Goriely, A.; Sargent, E.H.; Bakr, O.M. Automated Synthesis of Photovoltaic-Quality Colloidal Quantum Dots Using Separate Nucleation and Growth Stages. ACS Nano 2013, 7, 10158–10166. [Google Scholar] [CrossRef] [PubMed]
- Dransfield, G.P.; Fothergill, K.A.; Egerton, T.A. The use of plasma synthesis and pigment coating technology to produce an yttria stabilised zirconia having superior properties. In Euro Ceramics; Terpstra, R.A., Metsalaar, R.M., Eds.; Elsevier Applied Science Publishers: London, UK, 1989; Volume 1, pp. 275–279. [Google Scholar]
- Blackburn, S.R.; Egerton, T.A.; Jones, A.G. Vapour phase synthesis of nitride ceramic powders using a DC plasma. Br. Ceram. Proc. 1991, 47, 87–94. [Google Scholar]
- Fauchais, P.; Bourdin, E.; Coudert, J.F.; McPherson, R. High-pressure plasmas and their application to ceramic technology. In Topics in Current Chemistry; Boschke, F.L., Ed.; Springer: Berlin, Germany, 1983; Volume 107, pp. 59–184. [Google Scholar]
- Ross, F.M.; Tersoff, J.; Tromp, R.M. Coarsening of Self-Assembled Ge Quantum Dots on Si(001). Phys. Rev. Lett. 1998, 80, 984–987. [Google Scholar] [CrossRef]
- Ross, F.M.; Tromp, R.M.; Reuter, M.C. Transition States between Pyramids and Domes During Ge/Si Island Growth. Science 1999, 286, 1931–1934. [Google Scholar] [CrossRef]
- Ostwald, W.Z. Studien über die Bildung und Umwandlung fester Körper. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Mantzaris, N.V. Liquid-phase synthesis of nanoparticles: Particle size distribution dynamics and control. Chem. Eng. Sci. 2005, 60, 4749–4770. [Google Scholar] [CrossRef]
- Lifshitz, M.; Slezov, V.V. Kinetics of Diffusive Decomposition of Supersaturated Solid Solutions. Zh. Eksp. Sov. Phys. JETP 1959, 8, 331–339. [Google Scholar]
- Lifshitz, M.; Slezov, V.V. The Kinetics of Precipitation from Supersaturated Solid Solutions. J. Phys. Chem. Solids 1961, 19, 35–50. [Google Scholar] [CrossRef]
- Wagner, C.Z. Theorie der Alterung von Niederschlägen durch Umlösen (Ostwald-Reifung). Z. Elektrochem. 1961, 65, 581–591. [Google Scholar]
- Voorhees, P.W. Ostwald ripening of two-phase mixtures. Ann. Rev. Mater. Sci. 1992, 22, 197–215. [Google Scholar] [CrossRef]
- Porter, D.A.; Easterling, K.E. Phase Transformations in Metals and Alloys; Chapman and Hall: New York, NY, USA, 1992. [Google Scholar]
- Zinke-Allmang, M.; Feldman, L.C.; Grabow, M.H. Clustering on surfaces. Surf. Sci. Rep. 1992, 16, 377–463. [Google Scholar] [CrossRef]
- Taylor, P. Ostwald ripening in emulsions: Estimation of solution thermodynamics of the disperse phase. Adv. Colloid Interface Sci. 2003, 106, 261–285. [Google Scholar] [CrossRef]
- Bartelt, N.C.; Theis, W.; Tromp, R.M. Ostwald ripening of two-dimensional islands on Si(001). Phys. Rev. B 1996, 54, 11741–11751. [Google Scholar] [CrossRef]
- Goldfarb, I.; Briggs, G.A.D. Advances in germanium-silicon heteroepitaxy. Recent Res. Dev. Mater. Sci. 1998, 1, 189–213. [Google Scholar]
- Tromp, R.M.; Ross, F.M.; Reuter, M.C. Instability-driven SiGe island growth. Phys. Rev. Lett. 2000, 84, 4641–4644. [Google Scholar] [CrossRef]
- Lee, D.-K.; Park, S.-I.; Lee, J.K.; Hwang, N.-M. A theoretical model for digestive ripening. Acta Mater. 2007, 55, 5281–5288. [Google Scholar] [CrossRef]
- Lee, D.-K.; Hwang, N.-M. Thermodynamics and kinetics of monodisperse alloy nanoparticles synthesized through digestive ripening. Scr. Mater. 2009, 61, 304–307. [Google Scholar] [CrossRef]
- Rizza, C.C.; Strobel, M.; Heinig, K.H.; Bernas, H. Ion irradiation of gold inclusions in SiO2: Experimental evidence for inverse Ostwald ripening. Nucl. Instrum. Methods Phys. Res. B 2001, 178, 78–83. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Haynes, T.E.; Woody White, C.; MoberlyChan, W.J.; Roorda, S.; Aziz, M.J. Synthesis of nearly monodisperse embedded nanoparticles by separating nucleation and growth in ion implantation. Nano Lett. 2005, 5, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Rizza, G.; Cheverry, H.; Gacoin, T.; Lamasson, A.; Henry, S. Ion beam irradiation of embedded nanoparticles: Toward an in situ control of size and spatial distribution. J. Appl. Phys. 2007, 101, 014321. [Google Scholar] [CrossRef]
- Clark, M.D.; Kumar, S.K.; Owen, J.S.; Chan, E.M. Focusing Nanocrystal Size Distributions via Production Control. Nano Lett. 2011, 11, 1976–1980. [Google Scholar] [CrossRef]
- Clark, M.D. Growth laws for surfactant-coated nanocrystals: Ostwald ripening and size focusing. J. Nanopart. Res. 2014, 16, 2264. [Google Scholar] [CrossRef]
- Lu, A.H.; Salabas, E.L.; Schueth, F. Magnetic Nanoparticles: Synthesis, Protection, Functionalization, and Application. Angew. Chem. Int. Ed. 2007, 46, 1222–1244. [Google Scholar] [CrossRef]
- Kumar, S.; Gradzielski, M.; Mehta, S.K. The critical role of surfactants towards CdS nanoparticles: Synthesis, stability, optical and PL emission properties. RSC Adv. 2013, 3, 2662–2676. [Google Scholar] [CrossRef] [Green Version]
- Cookson, J. The preparation of palladium nanoparticles. Platin. Met. Rev. 2012, 56, 83–98. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murph, C.J. Seed-mediated growth approach for shape-controlled synthesis of spheroidal and rod-like gold nanoparticles using a surfactant template. Adv. Mater. 2001, 13, 1389–1393. [Google Scholar] [CrossRef]
- John, V.T.; Simmons, B.; McPherson, G.L.; Bose, A. Application and Characterization of Surfactants. Curr. Opin. Colloid Interface Sci. 2002, 7, 288–295. [Google Scholar] [CrossRef]
- Bao, Y.; An, W.; Turner, C.H.; Krishnan, K.M. The Critical Role of Surfactants in the Growth of Cobalt Nanoparticles. Langmuir 2009, 26, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Chebil, A.; Desbrieres, J.; Nouvel, C.; Six, J.-L.; Durand, A. Ostwald ripening of nanoemulsions stopped by combined interfacial adsorptions of molecular and macromolecular nonionic stabilizers. Colloid Surf. A Physicochem. Eng. Asp. 2013, 425, 24–30. [Google Scholar] [CrossRef]
- Lindfors, L.; Skantze, P.; Skantze, U.; Rasmusson, M.; Zackrisson, A.; Olsson, U. Amorphous Drug Nanosuspensions. 1. Inhibition of Ostwald Ripening. Langmuir 2006, 22, 906–910. [Google Scholar] [CrossRef]
- Pham, B.T.T.; Zondanos, H.; Such, C.H.; Warr, G.G.; Hawkett, B.S. Miniemulsion Polymerization with Arrested Ostwald Ripening Stabilized by Amphiphilic RAFT Copolymers. Macromolecules 2010, 43, 7950–7957. [Google Scholar] [CrossRef]
- Gupta, S.; Narayan, J. Selective Liquid-Phase Regrowth of Reduced Graphene Oxide, Nanodiamond, and Nanoscale Q-Carbon by Pulsed Laser Annealing for Radiofrequency Devices. ACS Appl. Nano Mater. 2020, 3, 5178–5188. [Google Scholar] [CrossRef]
- Parravicini, G.B.; Stella, A.; Ghigna, P.; Spinolo, G.; Migliori, A.; d’Acapito, F.; Kofman, R. Extreme undercooling (down to 90K) of liquid metal nanoparticles. Appl. Phys. Lett. 2006, 89, 033123. [Google Scholar] [CrossRef]
- Gupta, S.; Sachan, R.; Bhaumik, A.; Pant, P.; Narayan, J. Undercooling driven growth of Q-carbon, diamond, and graphite. MRS Commun. 2018, 8, 533–540. [Google Scholar] [CrossRef]
- Krishna, H.; Sachan, R.; Strader, J.; Favazza, C.; Khenner, M.; Kalyanaraman, R. Thickness-dependent spontaneous dewetting morphology of ultrathin Ag films. Nanotechnology 2010, 21, 155601. [Google Scholar] [CrossRef]
- Yang, S.; Xu, F.; Ostendorp, S.; Wilde, G.; Zhao, H.; Lei, Y. Template-Confined Dewetting Process to Surface Nanopatterns: Fabrication, Structural Tunability, and Structure-Related Properties. Adv. Funct. Mater. 2011, 21, 2446–2455. [Google Scholar] [CrossRef]
- Watt, J.; Cheong, S.; Tilley, R.D. How to control the shape of metal nanostructures in organic solution phase synthesis for plasmonics and catalysis. Nano Today 2013, 8, 198–215. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, C.; Huang, H.; Zhang, L.; Jiang, Z.; Chen, G.; Jia, Y.; Kuang, Q.; Xie, Z.; Zheng, L. Surfactant-Concentration-Dependent Shape Evolution of Au–Pd Alloy Nanocrystals from Rhombic Dodecahedron to Trisoctahedron and Hexoctahedron. Small 2013, 9, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Y.; Tao, F.; Wang, L. Electrochemical Fabrication of Shape-Controlled Copper Hierarchical Structures Assisted by Surfactants. J. Nanomater. 2012, 2012, 6. [Google Scholar] [CrossRef]
- Xing, R.; Lehmler, H.-J.; Knutson, B.L.; Rankin, S. Demixed Micelle Morphology Control in Hydrocarbon/Huorocarbon Cationic Surfactant Templating of Mesoporous Silica. J. Phys. Chem. C 2010, 114, 17390–17400. [Google Scholar] [CrossRef]
- Xiao, J.; Qi, L. Surfactant-assisted, shape-controlled synthesis of gold nanocrystals. Nanoscale 2011, 3, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Qi, L. Low-Temperature Synthesis of Star-Shaped PbS Nanocrystals in Aqueous Solutions of Mixed Cationic/Anionic Surfactants. Adv. Mater. 2006, 18, 359–362. [Google Scholar] [CrossRef]
- Burlakov, V.M.; Hassan, Y.; Danaie, M.; Snaith, H.J.; Goriely, A. Competitive Nucleation Mechanism for CsPbBr3 Perovskite Nanoplatelets Growth. J. Phys. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Burlakov, V.M.; Goriely, A. Thermodynamic limit for particle monodispersity: How narrow can a particle size distribution be? EPL 2017, 119, 50001. [Google Scholar] [CrossRef]
- Donohue, M.D.; Prausnitz, J.M. Combinatorial entropy of mixing molecules that differ in size and shape. A simple approximation for binary and multicomponent mixtures. Can. J. Chem. 1975, 53, 1586–1594. [Google Scholar] [CrossRef]
- Battaile, C.C. The Kinetic Monte Carlo method: Foundation, implementation, and application. Comput. Methods Appl. Mech. Eng. 2007, 197, 3386–3398. [Google Scholar] [CrossRef]
- Uppenbrink, J.; Wales, D.J. Structure and energetics of model metal clusters. J. Chem. Phys. 1992, 96, 8520–8534. [Google Scholar] [CrossRef]
- Jortner, J. Cluster size effects. Z. Phys. D 1992, 24, 247–275. [Google Scholar] [CrossRef]
- Lordeiro, R.A.; Guimaraes, F.F.; Belchior, J.C.; Johnston, R.L. Determination of main structural compositions of nanoalloy clusters of Cux Auy (x + y ≤ 30) using a genetic algorithm approach. Int. J. Quantum Chem. 2003, 95, 112–125. [Google Scholar] [CrossRef]
- Böyükata, M.J.; Belchior, C. Structural and energetic analysis of copper clusters: MD study of Cun (n = 2–45). J. Braz. Chem. Soc. 2008, 19, 884–893. [Google Scholar] [CrossRef] [Green Version]
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Robert, J.; Flatt, R.J.; Estrela-Lopis, I.; Llop, J.; et al. Nanoparticle decoration with surfactants: Molecular interactions, assembly, and applications. Surf. Sci. Rep. 2017, 72, 1–58. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlakov, V.M.; Goriely, A. Reverse Coarsening and the Control of Particle Size Distribution through Surfactant. Appl. Sci. 2020, 10, 5359. https://doi.org/10.3390/app10155359
Burlakov VM, Goriely A. Reverse Coarsening and the Control of Particle Size Distribution through Surfactant. Applied Sciences. 2020; 10(15):5359. https://doi.org/10.3390/app10155359
Chicago/Turabian StyleBurlakov, Victor M., and Alain Goriely. 2020. "Reverse Coarsening and the Control of Particle Size Distribution through Surfactant" Applied Sciences 10, no. 15: 5359. https://doi.org/10.3390/app10155359
APA StyleBurlakov, V. M., & Goriely, A. (2020). Reverse Coarsening and the Control of Particle Size Distribution through Surfactant. Applied Sciences, 10(15), 5359. https://doi.org/10.3390/app10155359