Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
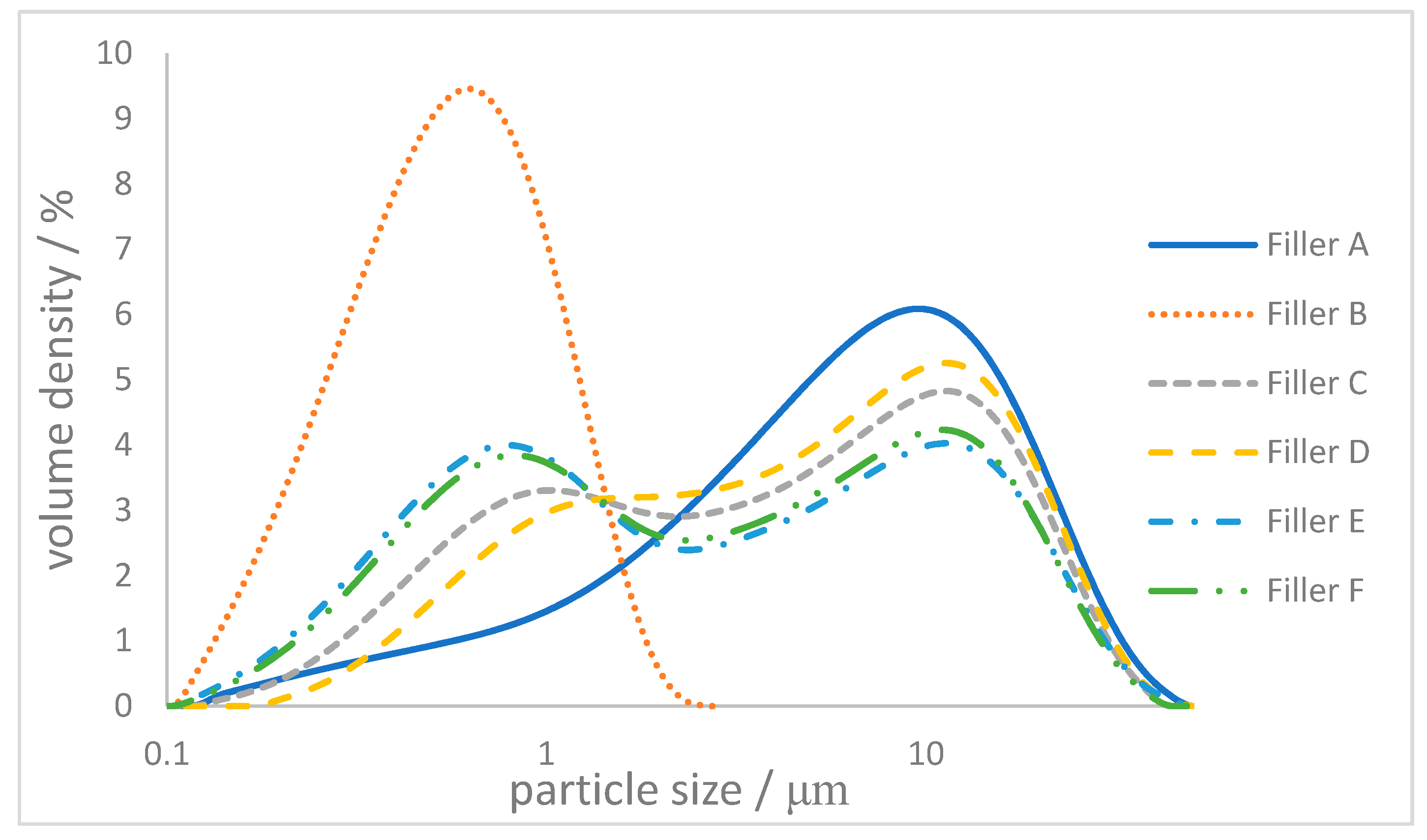
2.2. Controlling the Particle Size Distribution
2.3. Formulating Filler and Polyamide 12
2.4. Incorporating the Fillers into the Polyamide 12 by Compounding
2.5. Analytical Thermal Methods Applied to Composites
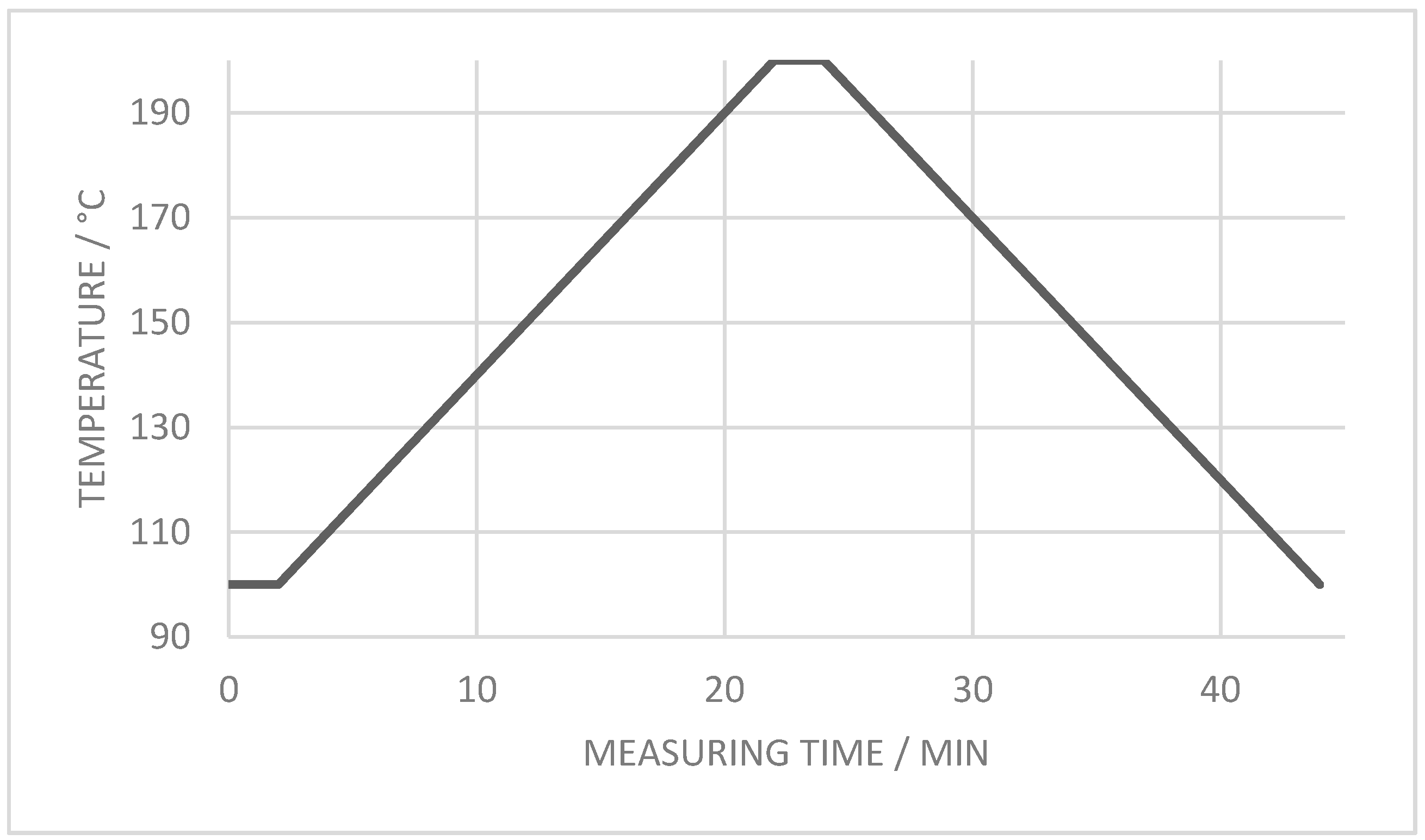
2.5.1. Thermogravimetric Analysis
2.5.2. Melt Flow Index
2.5.3. Differential Scanning Calorimetry
2.5.4. Scanning Electron Microscopy
3. Results and Discussion
3.1. Filler Distribution Homogeneity
3.2. Thermal Properties
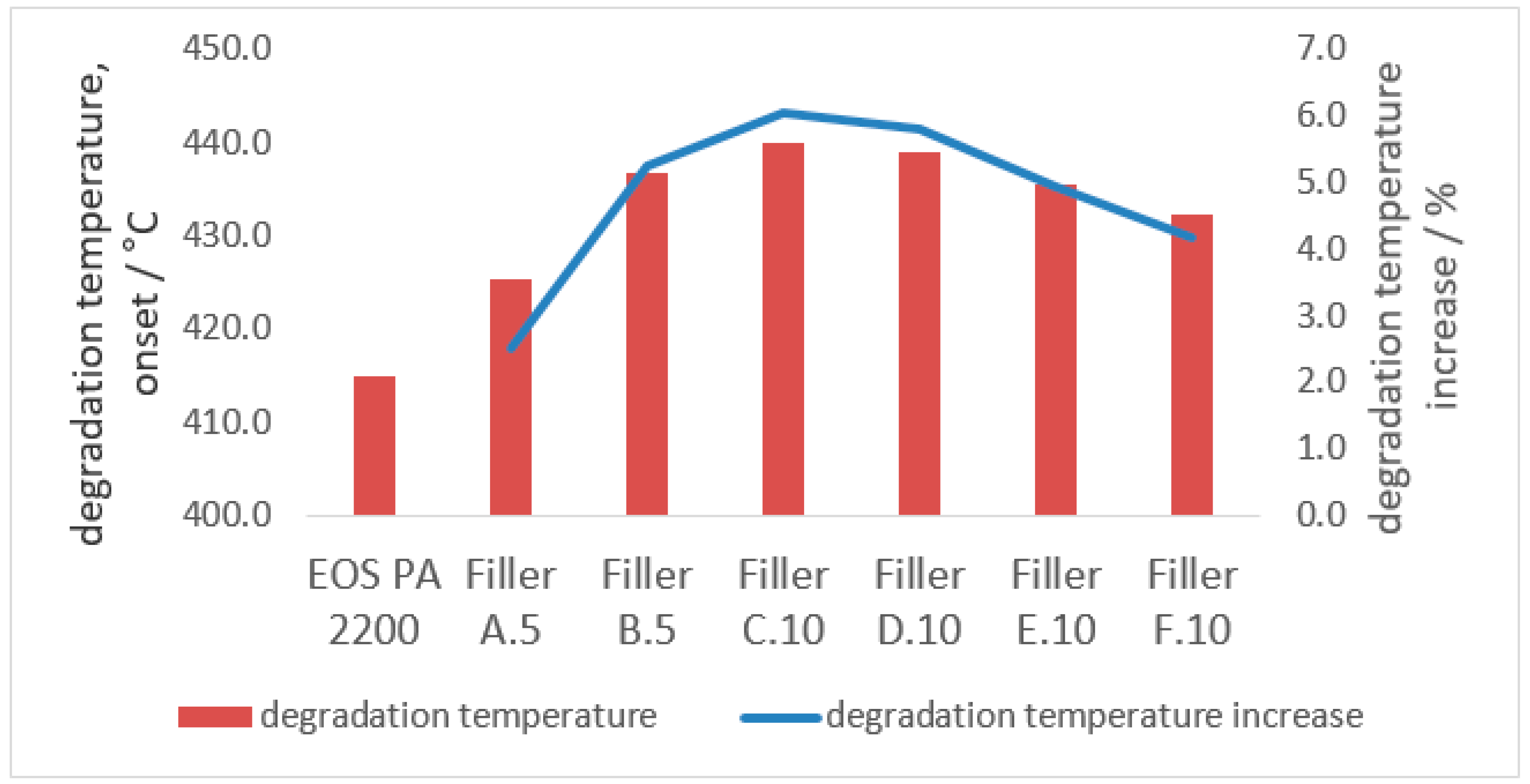
3.2.1. Degradation Temperature
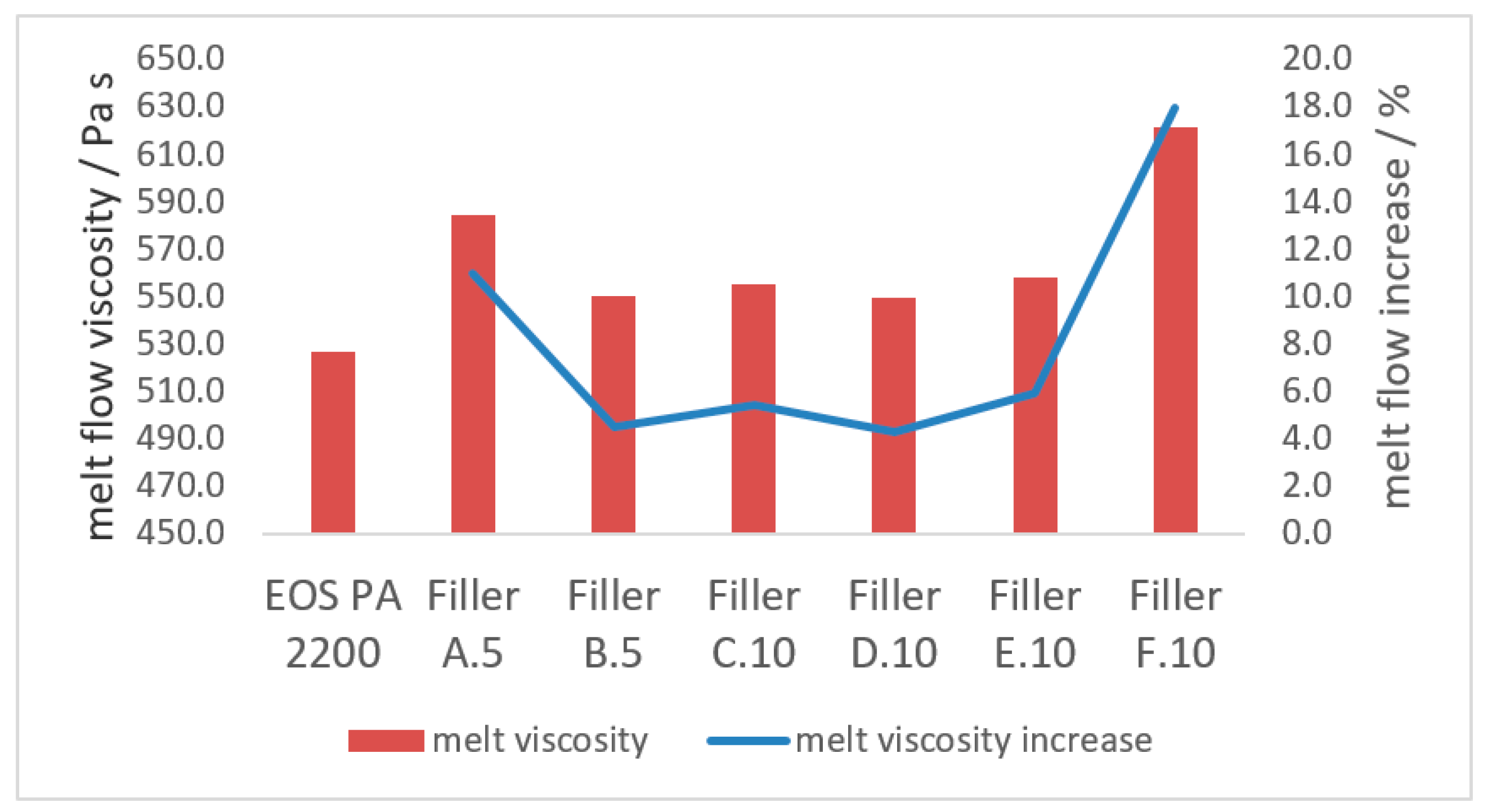
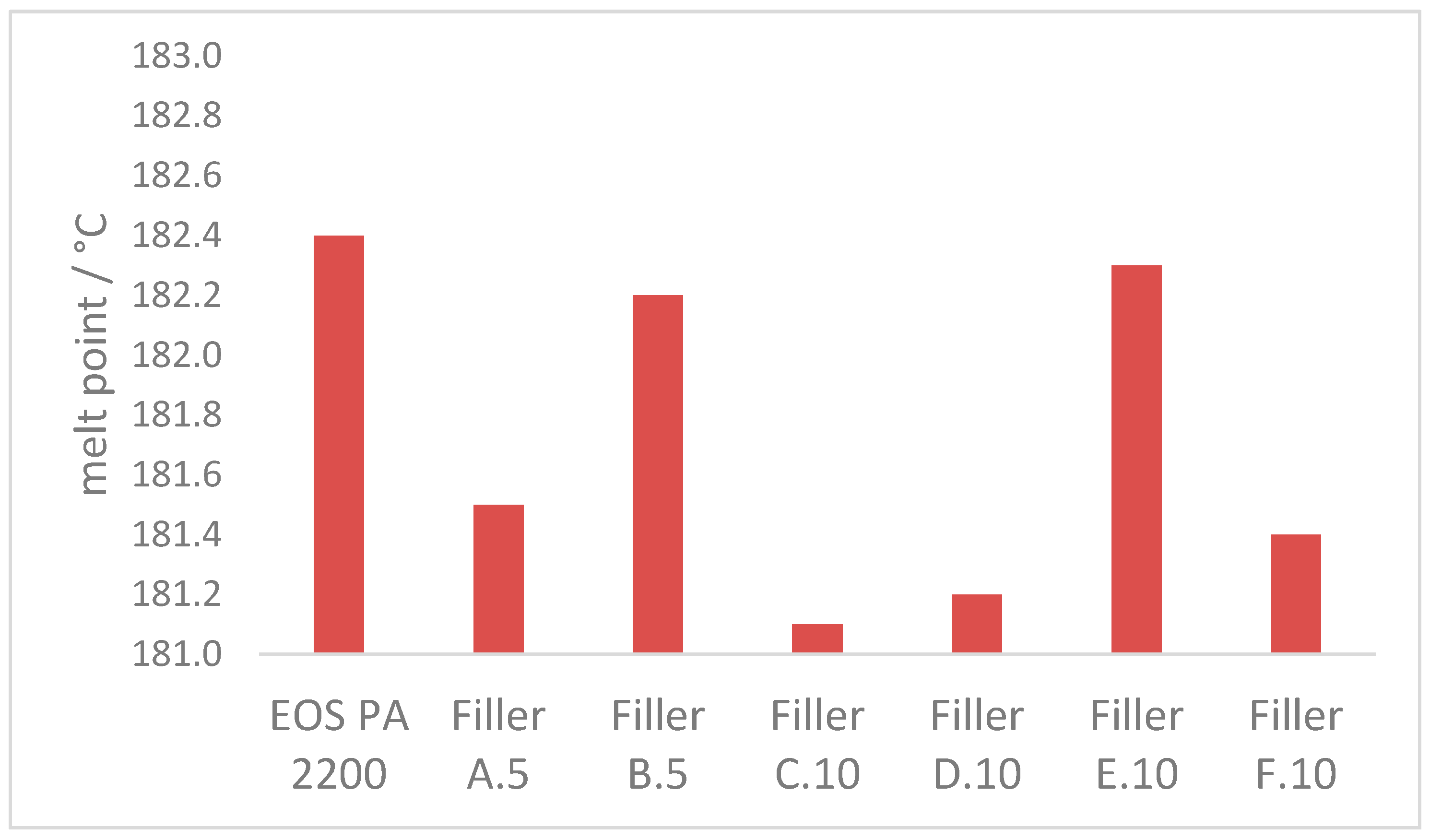
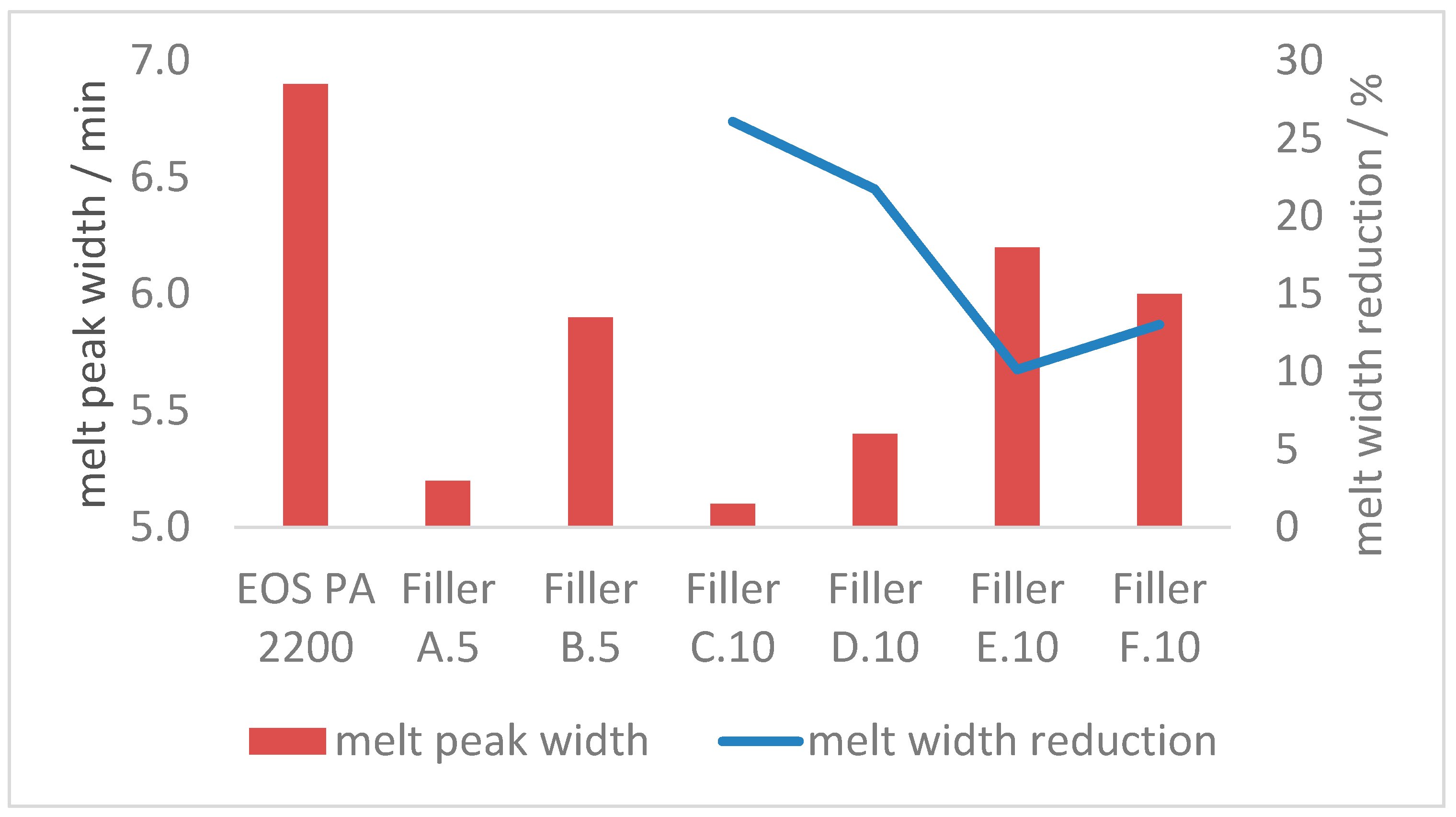
3.2.2. Melt Properties
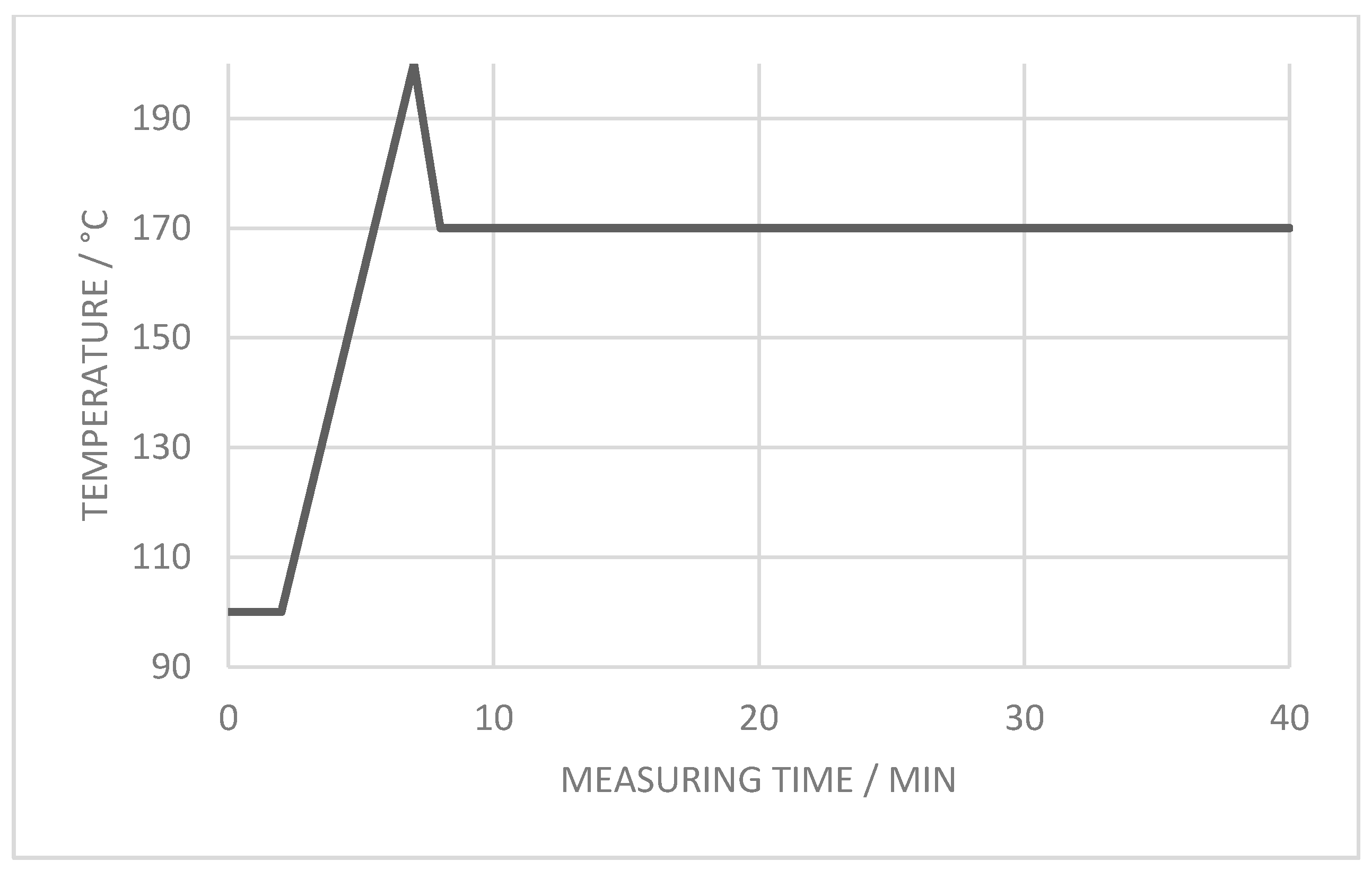
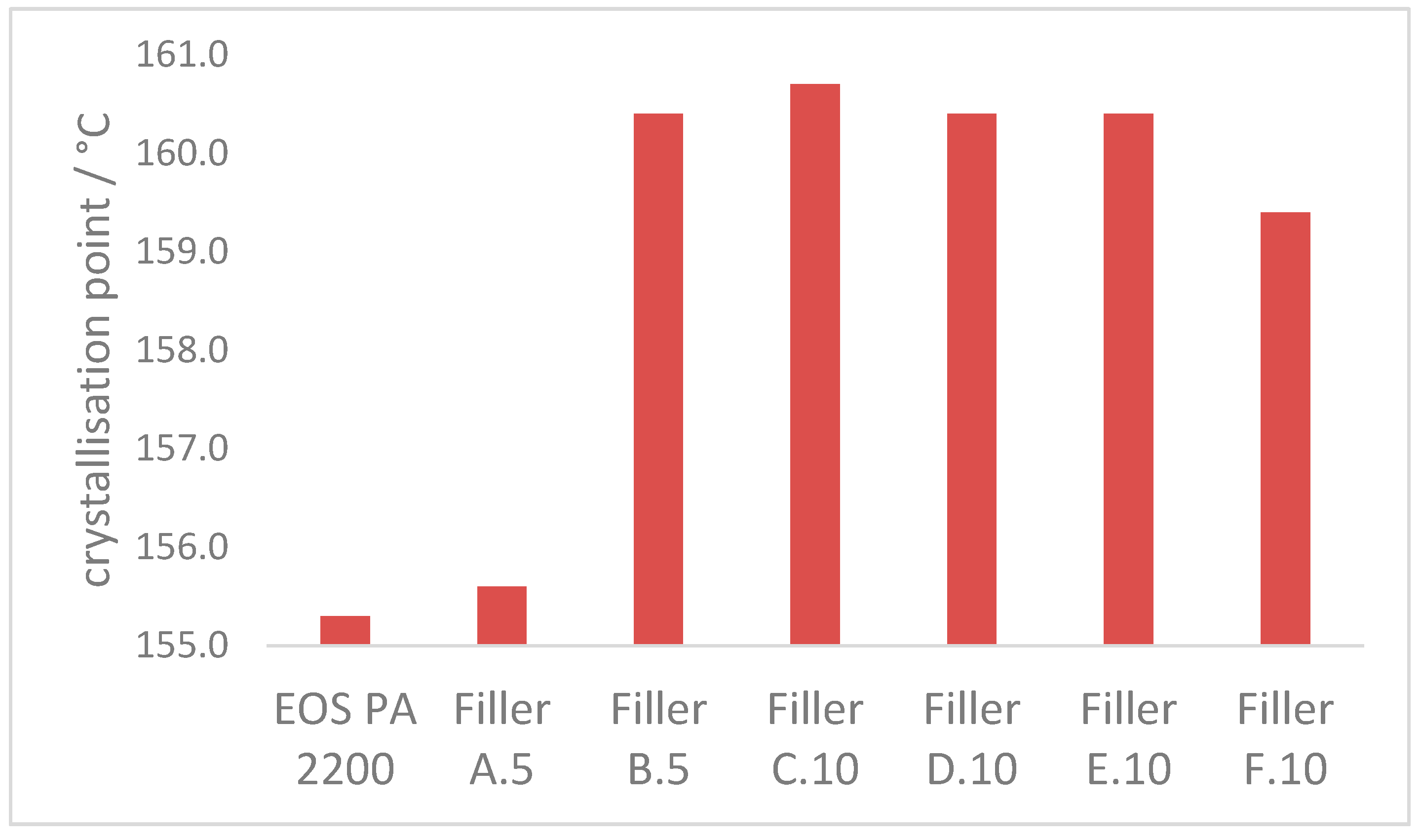
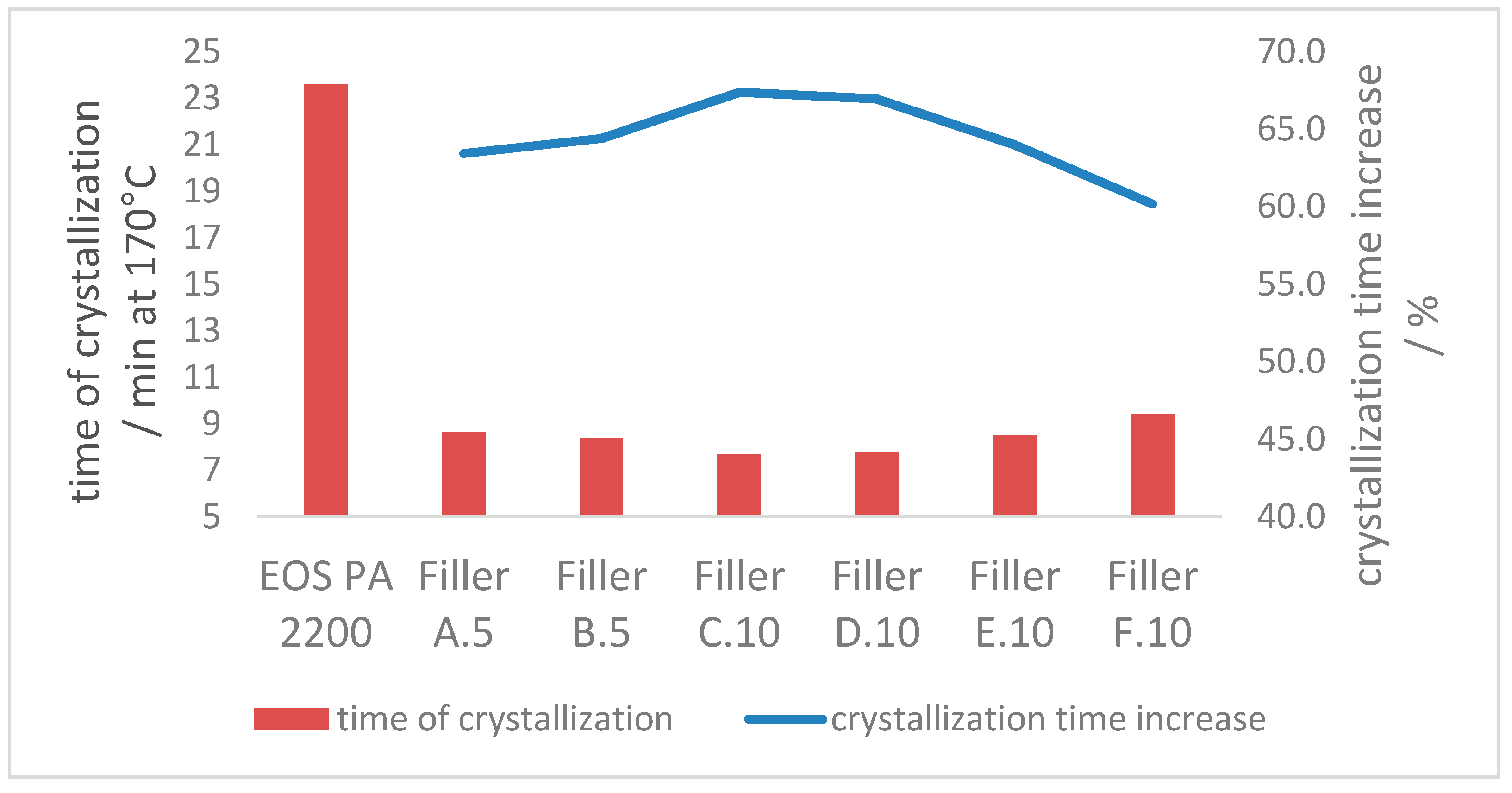
3.2.3. Crystallization Properties
4. Conclusions
- The increase of the melt flow, due to the introduction of a coarse filler fraction, can be reduced with the introduction of a finer filler into the blend. This increase in melt viscosity generated by the coarse filler can be reduced up to 60% with the precise amount of fine filler in the blend. If the fine filler fraction exceeds the optimum range of 80–130 m2 per 100 g polyamide 12, the improvement on the melt viscosity is reduced or even lost totally;
- The degradation temperature of the polymer compound can be increased by up to 6% with the implementation of at least 10 m2 of the coarse and 80 m2 of the fine filler fraction within 100 g of polyamide 12;
- With the combination of 10 m2 of coarse and 130 m2 fine filler in 100 g of polyamide 12, the melting transition can be sharpened up to 25% (reduced time required);
- Due to the higher single-particle-volume of the coarse fraction, the effect on the overall degradation and melting properties is reduced if the coarse filler fraction falls below the optimum threshold of 10 m2 per 100 g polyamide 12. The same counts for when the finer filler fraction is increased significantly;
- The crystallization process at 170 °C can be accelerated approx. 60% with the introduction of enough additional nucleation points, independent of the ratio of coarse and fine filler material in the blend. The crystallization point is significantly increased by the higher particle number of the fine filler fraction, in comparison with the coarse filler introduction only.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ippolito, F.; Rentsch, S.; Hübner, G.; Claypole, T.; Gane, P. Influence of calcium carbonate on polyamide 12 regarding melting, formability and crystallization properties. Compos. Part B Eng. 2019, 164, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Mueller, B. Additive manufacturing technologies–rapid prototyping to direct digital manufacturing. Assem. Autom. 2012, 32. [Google Scholar] [CrossRef]
- Hague, R.; Mansour, S.; Saleh, N. Design opportunities with rapid manufacturing. Assem. Autom. 2003, 23, 346–356. [Google Scholar] [CrossRef]
- Gibson, I.; Rosen, D.; Stucker, B. Additive Manufacturing Technologies: 3D Printing, Rapid Prototyping, and Direct Digital Manufacturing; Springer: New York, NY, USA, 2014. [Google Scholar]
- Kumar, S. Selective Laser Sintering/Melting, Comprehensive Materials Processing; Hashmi, S., Batalha, G.F., Van Tyne, C.J., Yilbas, B., Eds.; Elsevier: Oxford, UK, 2014; pp. 93–134. [Google Scholar]
- Lin, L.-L.; Shi, Y.-S.; Zeng, F.-D.; Huang, S.-H. Microstructure of selective laser sintered polyamide. J. Wuhan Univ. Technol. Sci. Ed. 2003, 18, 60–63. [Google Scholar]
- Childs, T.H.C.; Berzins, M.; Ryder, G.R.; Tontowi, A. Selective laser sintering of an amorphous polymer—simulations and experiments. Proc. Inst. Mech. Eng. Part B J. Eng. Manuf. 1999, 213, 333–349. [Google Scholar] [CrossRef]
- Schmidt, M.; Pohle, D.; Rechtenwald, T. Rechtenwald, Selective laser sintering of PEEK. CIRP Ann. 2007, 56, 205–208. [Google Scholar] [CrossRef]
- Koo, J.; Lao, S.; Ho, W.; Ngyuen, K.; Cheng, J.; Pilato, L.; Wissler, G.; Ervin, M. Polyamide nanocomposites for selective laser sintering. Proc. SFF Symp. Austin 2006, 392–409. [Google Scholar]
- Schmid, M.; Amado, A.; Wegener, K. Polymer Powders for Selective Laser Sintering (SLS); ETH-Zürich: Zürich, Switzerland, 2014. [Google Scholar]
- Kim, J.; Creasy, T. Selective laser sintering characteristics of nylon 6/clay-reinforced nanocomposite. Polym. Test. 2004, 23, 629–636. [Google Scholar] [CrossRef]
- Goodridge, R.; Tuck, C.; Hague, R. Laser sintering of polyamides and other polymers. Prog. Mater. Sci. 2012, 57, 229–267. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, K.; Bourell, D.L. Preparation and laser sintering of limestone PA 12 composite. Polym. Test. 2014, 37, 210–215. [Google Scholar] [CrossRef]
- Ippolito, F.; Hübner, G.; Claypole, T.; Gane, P.A.C. Influence of the Surface Modification of Calcium Carbonate on Polyamide 12 Composites. Polymers 2020, 12, 1295. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, R.; Goel, A.; Bourell, D. Sustainability issues in laser-based additive manufacturing. Phys. Procedia 2010, 5, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Ford, S.J.; Despeisse, M. Additive manufacturing and sustainability: An exploratory study of the advantages and challenges. J. Clean. Prod. 2016, 137, 1573–1587. [Google Scholar] [CrossRef]
- Le Bourhis, F.; Kerbrat, O.; Hascoet, J.-Y.; Mognol, P. Sustainable manufacturing: Evaluation and modeling of environmental impacts in additive manufacturing. Int. J. Adv. Manuf. Technol. 2013, 69, 1927–1939. [Google Scholar] [CrossRef] [Green Version]
- Kruth, J.-P.; Levy, G.; Schindel, R.; Craeghs, T.; Yasa, E. Consolidation of polymer powders by selective laser sintering. In Proceedings of the 3rd International Conference on Polymers and Moulds Innovations, Ghent, Belgium, 19–21 September 2008; pp. 15–30. [Google Scholar]
- Höhne, G.W.H.; Hemminger, W.; Flammersheim, H.-J. Theoretical Fundamentals of Differential Scanning Calorimeters, Differential Scanning Calorimetry; Springer: Berlin/Heidelberg, Germany, 1996; pp. 21–40. [Google Scholar]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Drummer, D.; Rietzel, D.; Kühnlein, F. Development of a characterization approach for the sintering behavior of new thermoplastics for selective laser sintering. Phys. Procedia 2010, 5, 533–542. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omyacarb 10-AV | PA2200 | |
|---|---|---|
| Producer/supplier | Omya International | EOS e-Manufacturing |
| Volume-based median Particle size, dv50 | 9 μm | 60 μm |
| Particle shape | irregular | spherical |
| Approx. thermal Conductivity at 298 K | 1.3 Wm−1 K−1 | 0.2 Wm−1 K−1 |
| Approx. specific heat | 0.8 kJkg−1 K−1 | 1.2 kJkg−1 K−1 |
| Filler | dv10/μm | dv50/μm | dv90/μm | Specific Surface Area/m2g−1 |
|---|---|---|---|---|
| A | 0.99 | 6.34 | 18.9 | 1.9 |
| B | 0.24 | 0.56 | 1.16 | 23.3 |
| Filler | dv50/μm | Amount Filler A/w/w% | Amount Filler B/w/w% | Specific Surface Area/m2g−1 |
|---|---|---|---|---|
| C | 4.12 | 50 | 50 | 12.2 |
| D | 5.00 | 70 | 30 | 7.8 |
| E | 2.59 | 30 | 70 | 21.8 |
| F | 2.87 | 33 | 66 | 21.6 |
| Filler Definition | Used Filler | Filler Amount/w/w% | Total Carbonate Surface per 100 g Polyamide 12/m2 | Filler A Surface per 100 g Polyamide 12/m2 | Filler B Surface per 100 g Polyamide 12/m2 |
|---|---|---|---|---|---|
| A.5 | A | 5 | 10 | 10 | 0 |
| B.5 | B | 5 | 120 | 0 | 120 |
| C.10 | C | 10 | 141 | 11 | 130 |
| D.10 | D | 10 | 95 | 15 | 80 |
| E.10 | E | 10 | 187 | 6 | 181 |
| F.10 | F | 10 | 286 | 11 | 275 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ippolito, F.; Hübner, G.; Claypole, T.; Gane, P. Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12. Appl. Sci. 2021, 11, 641. https://doi.org/10.3390/app11020641
Ippolito F, Hübner G, Claypole T, Gane P. Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12. Applied Sciences. 2021; 11(2):641. https://doi.org/10.3390/app11020641
Chicago/Turabian StyleIppolito, Fabio, Gunter Hübner, Tim Claypole, and Patrick Gane. 2021. "Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12" Applied Sciences 11, no. 2: 641. https://doi.org/10.3390/app11020641
APA StyleIppolito, F., Hübner, G., Claypole, T., & Gane, P. (2021). Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12. Applied Sciences, 11(2), 641. https://doi.org/10.3390/app11020641