1. Introduction
Modeling light—metal interaction for ultrafast laser fields remains a challenging issue as the role of transient electronic response in the metal band structure is a dominant factor in optical properties. Intense photoexcitation in the femtosecond range for fluence near the ablation threshold, typically 0.2 J/cm
for most of transition metals, forces conduction electrons in metals to move around the lattice ions, enabling photon absorption through an inverse bremsstrahlung process [
1]. Beyond this classical view, electrons undergo electron-electron and electron-phonon scattering, resulting in a photon absorption process as well as an internal thermalization [
2,
3]. Throughout the laser-solid absorption, the electrons are redistributed within the density of states respecting the Pauli exclusion principle. This involves intraband and interband displacements of the electron population, followed by thermalization within the bands [
4]. Screening effects determine a subsequent evolution of the bands themselves [
5]. The evolution of optical properties relies directly on the transient electronic band structure. The prediction of optical properties thus requires an inaccessible kinetic description, challenging the rationalization of the collective processes. To interpret and predict the optical dynamics in time-dependent ultrashort laser—metal modeling, clarification and simplification of the absorption mechanisms are required. To achieve this, tractable models and simulation approaches capable of describing fast electron dynamics are formulated.
For pure transition metals irradiated in IR-visible range, the absorption length corresponding to the optical skin depth is on a scale of a few tens of nanometers, and the optical properties are determined by the bulk properties. For irradiated quasi-flat and rough surfaces, this strong energy confinement is able to trigger a plethora of surface ultrafast phenomena at the nanoscale [
6,
7]. The observed material reflectivity and thus the amount of laser energy absorbed in the skin depth is defined by the collective electronic response to the laser field through intraband and interband transitions in sub-bands of the crystal state [
8,
9,
10,
11,
12]. The time-resolved complex refractive index of the laser-irradiated surface has been previously reported by a dual-angle reflectometry technique, which was applied to study the optical dynamics of
d-band electrons in transition metals near the threshold fluence [
13,
14,
15,
16]. To better determine the refractive index without Fresnel equations, a direct measurement of optical properties has been recently proposed by an ultrafast pump-probe method combining ellipsometry with reflectometry [
17,
18]. Recent self-reflectivity measurements of femtosecond-laser-heated aluminum and tungsten also confirm the importance of the transient density of states for interpreting the experimental data [
19]. Moreover, both time-resolved and integrated optical data showed a strongly increased optical coupling due to the fast variation of the optical properties during irradiation with ultrashort laser pulses.
Fitting these optical properties on a relatively large spectrum is of great interest for spectroscopy, plasmonics, nanophotonics, and optical engineering, which requires dispersion models, such as Drude, Drude-Lorentz, Critical Points, or Partial-Fraction [
20,
21,
22]. The goal of the present work is to propose a straightforward Drude-Lorentz model with electron-phonon nonequilibrium parameters that ultimately aims at being implemented into numerical models as Maxwell solvers. Our approach provides fitting values of optical properties during the nonequilibrium state lasting a few picoseconds when the lattice temperature remains undisturbed at 300 K, modifying the material primary response to the laser irradiation. A “hot” electron temperature range of 10,000–30,000 K was considered to obtain a non-negligible effect on permittivity and, at the same time, to work in the limits of material stability [
23]. Such temperatures are expected for the typical laser irradiation parameters: pulse duration 10–100 fs, fluence 0.1–0.2 J/cm
and wavelength 800 nm [
4,
19]. We assume that the lattice is at room temperature for generality and larger applicability to different nonequilibrium conditions even though an increase of 100–300 K can be expected within 100 fs. Such an increase is insignificant in the given study since the lattice temperature stays well below the melting threshold.
The nonequilibrium data were obtained using ab initio molecular dynamics and the Kubo-Greenwood formalism [
14,
24]. These calculations, combined with Kramers-Kronig relations, give access to the complex optical permittivities of the considered metals for several electron temperatures from 300 K to 30,000 K. They serve as reference points for the fitting process for real and imaginary parts of the permittivities, optimizing parameters such as oscillator and collision frequencies. (In their attempt to describe the interaction between atoms and electric fields in classical terms, Lorentz proposed that electrons are bound to the ions by a force that behaves as a spring, according to Hooke’s Law. In the so-called Lorentz oscillator model, the applied electric field interacting with the electron charge, causes stretching or compression of the spring, which sets the electron into oscillating motion.)
2. Methods
First, ab initio calculations of optical permittivity have been performed for different transition metals (Cr, W, Ti, Fe, Au, and Ni) at different electron temperatures up to 30,000 K using A
binit software [
25]. The electronic structure (density of states, band filling, electron distribution) were calculated using a finite-temperature density functional theory with appropriate exchange-correlation functionals (GGA for Cr, W, Ti, Fe, and Ni and LDA for Au) and projector-augmented wave atomic data to consider nuclei and core electrons effects. The finite electronic temperature
is taken into account by considering a Fermi–Dirac distribution function applied to the Kohn-Sham eigenstates, thus describing a
-dependent electronic structure as a result of self-consistent calculation. The quantum molecular dynamic simulation was performed at an ionic temperature of 300 K to account for electron-phonon interaction in optical properties. Then we used a Kubo-Greenwood formalism to describe the possible electronic transitions and Kramers-Kronig relation to finally calculate the permittivity. The high electron temperature is considered to reproduce the transient distribution of photoexcited electrons at the laser pulse fluences used for nanostructuring. A detailed description of these calculations can be found in our previous work [
24] with some additional information for Au in Ref. [
5].
Then, the Drude-Lorentz model was fitted to the ab initio permittivity with the aim of providing a simple and convenient way to interpret and apply the calculated data. The Drude-Lorentz model is often used for the parametrization of optical constants of metals [
20] with the complex permittivity dispersion describing the free electrons (intraband transitions) and bound electrons (interband transitions):
The intraband contribution
is described by the free-electron Drude model:
where
is the plasma frequency,
is the free-electron oscillator strength, and
is the damping constant related to the electron collision frequency. The interband contribution
follows the Lorentz model:
which describes
k interband transitions with frequency
, oscillator strength
, and lifetime
.
To fit the Drude-Lorentz model to the ab initio data, the Basin-hopping optimization method was used. The Basin-hopping algorithm was designed to mimic the energy minimization of clusters of atoms and thus is suitable for our case where the Drude-Lorentz oscillators have similar energy landscapes [
26]. The difference between the model and the data was minimized based on the objective function
:
where the sum runs over all
N data points, and
and
are the real and imaginary parts of the permittivity
. Note that the relative permittivity is considered everywhere. A reduced objective function
was also calculated and presented below. A value of
less than unity is expected for a good fit. A Python code implementing the data fitting is provided in the
Supplementary Materials.
During the optimization procedure, we fixed
in Equation (
3). The preliminary analysis showed that such a number of interband terms was sufficient for all considered metals and electron temperatures. For significant results and faster convergence, we also set the constraints on the optimization parameters guided by the physical sense and the values reported in [
20]. The plasma frequency in Equations (
2) and (
3) is related to the free-electron density
by the equation
. Note that the free-electron oscillator strength
is often included in the plasma frequency via the electron effective mass
. The free-electron density depends on the electron temperature
and was estimated from ab initio calculations using the procedure described in Ref. [
5]. Both
and
are reported in
Table 1 at different electron temperatures.
3. Results
The Drude-Lorentz model is mostly used to fit the experimental data for different metals and frequency intervals [
20,
27,
28]. Thus, we first made sure that the proposed algorithm can correctly fit the experimentally measured permittivities. The optimal values for the model parameters obtained for the experimental data tabulated in the
Handbook on Optical Constants of Metals (Adachi, 2012) [
29] are given in
Table 2 (
and
are in eV). The model was fitted in the 0.05–6 eV range with
interband oscillators and the plasma frequencies given in
Table 1 at 300 K.
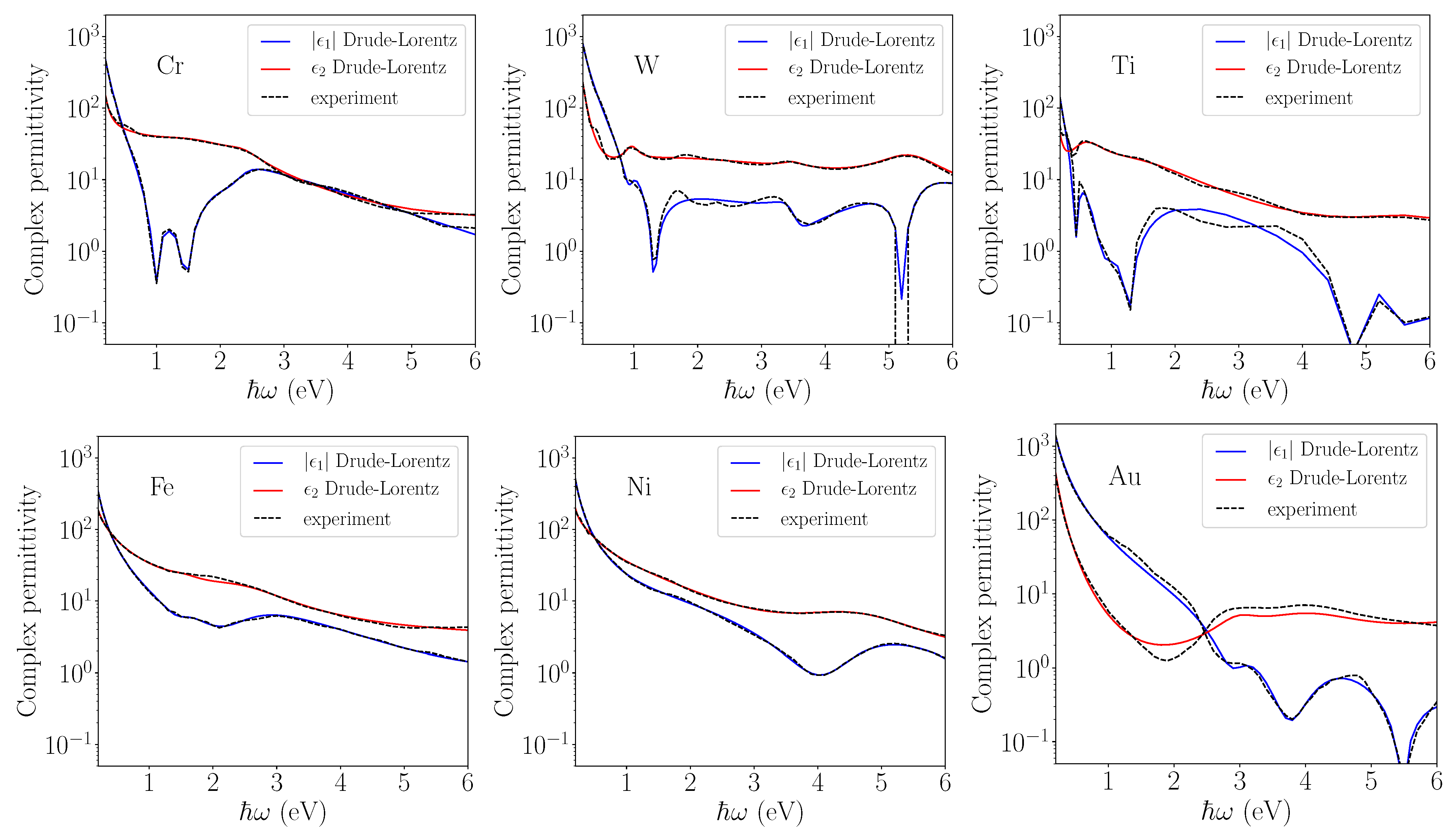
Figure 1 shows a general good agreement between our model and experimental data for all considered metals.
Table 1.
Free-electron densities and plasma frequencies for different metals at different electron temperatures .
Table 1.
Free-electron densities and plasma frequencies for different metals at different electron temperatures .
| Metal | (K) | ( cm) | (eV) |
|---|
| | 300 | 1.67 | 15.17 |
| Cr | 10,000 | 1.67 | 15.17 |
| | 25,000 | 1.67 | 15.17 |
| | 300 | 1.40 | 13.88 |
| W | 10,000 | 1.40 | 13.88 |
| | 25000 | 1.52 | 14.50 |
| | 300 | 0.79 | 10.45 |
| Ti | 10,000 | 0.79 | 10.45 |
| | 25,000 | 0.85 | 10.82 |
| | 300 | 1.11 | 12.36 |
| Fe | 15,000 | 1.28 | 13.28 |
| | 30,000 | 1.45 | 14.14 |
| | 300 | 1.42 | 13.98 |
| Au | 10,000 | 1.47 | 14.26 |
| | 25,000 | 1.65 | 15.10 |
| | 300 | 1.28 | 13.28 |
| Ni | 10,000 | 1.55 | 14.63 |
| | 25,000 | 1.83 | 15.87 |
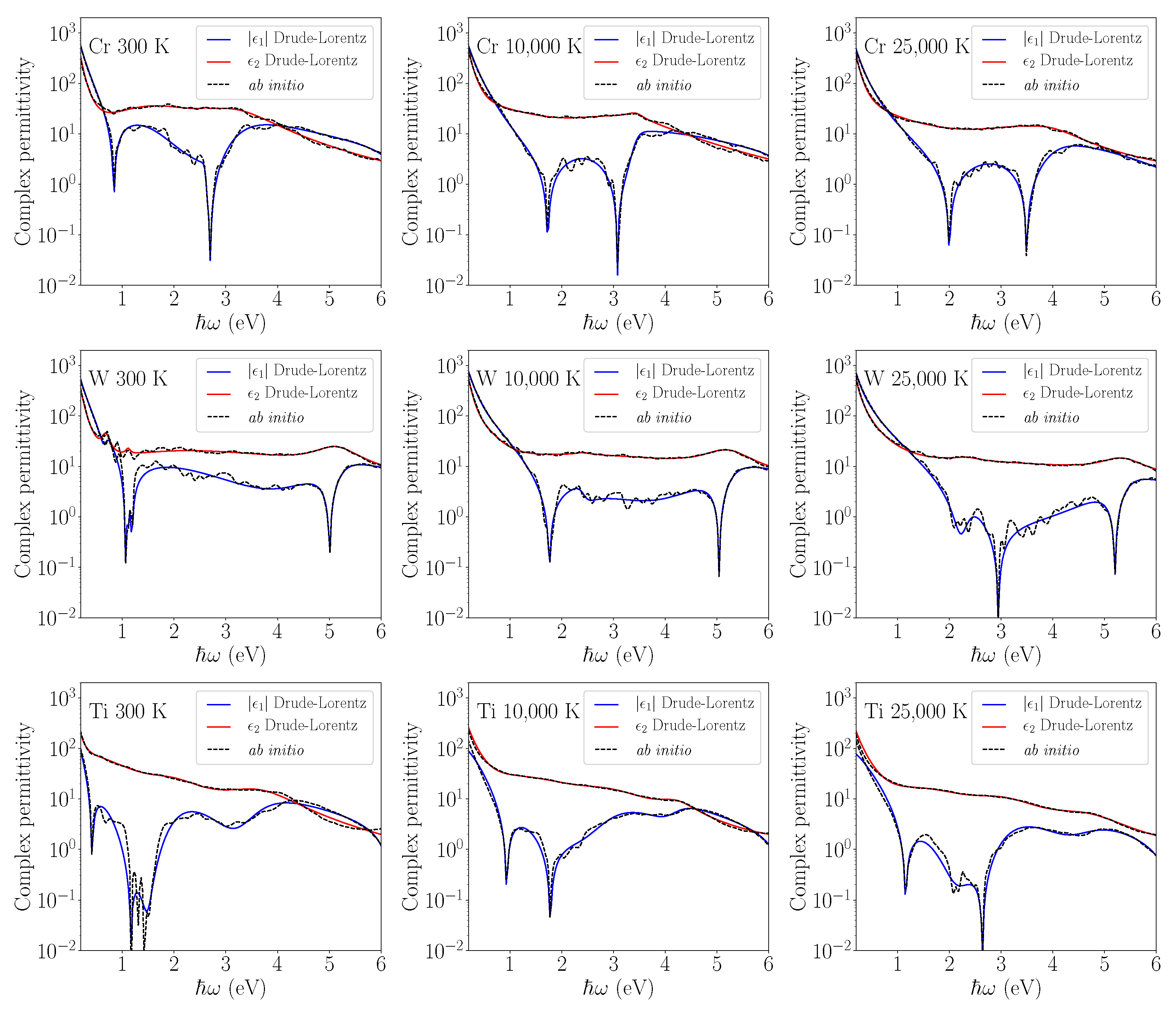
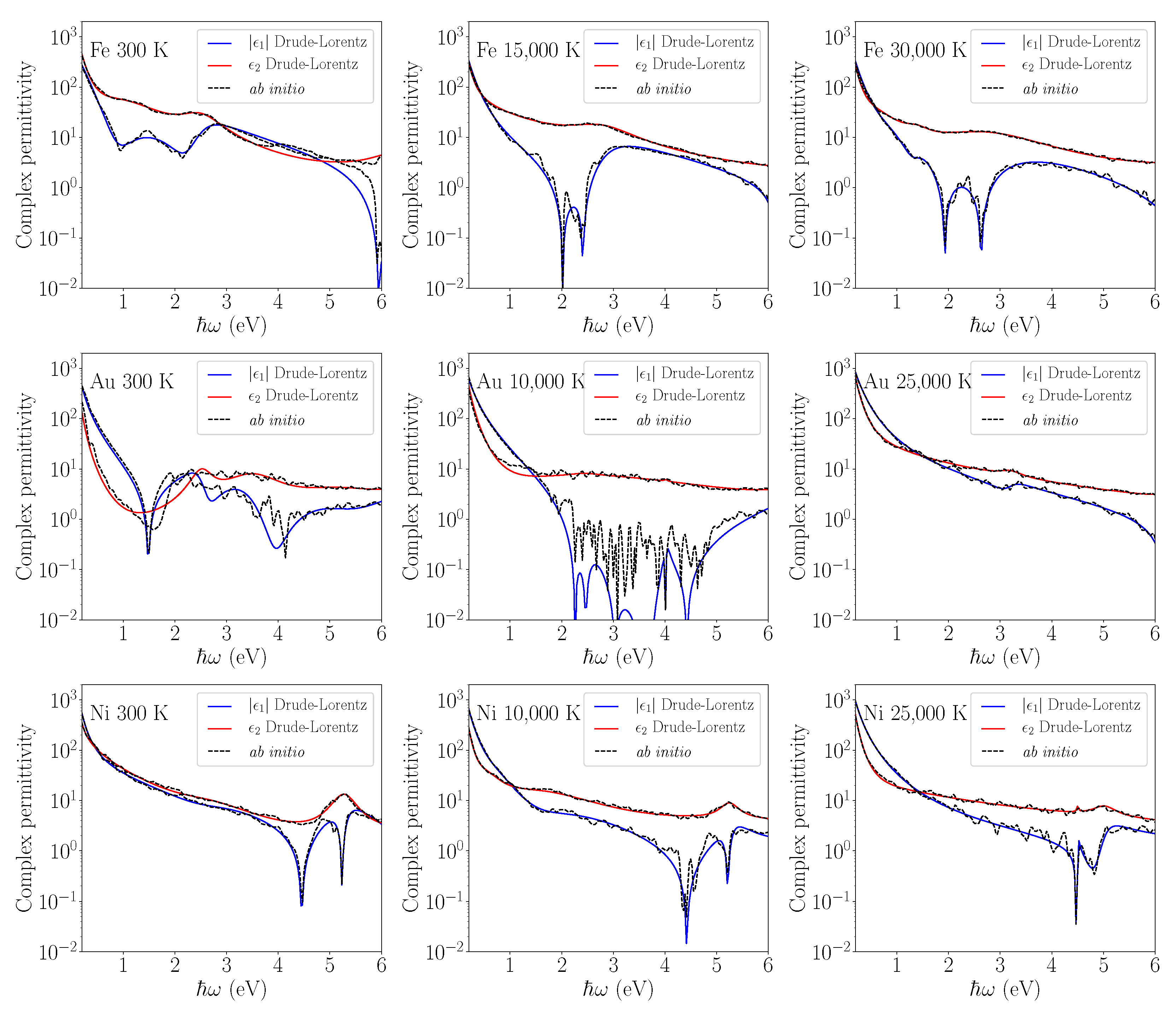
The results of ab initio calculations and the optimized Drude-Lorentz model at different electron temperatures are shown in
Figure 2 for Cr, W, and Ti and in
Figure 3 for Fe, Au, and Ni, with the values of the optimization parameters given in
Table 3 and
Table 4, respectively. The fitting range 0.05–6 eV was chosen to correspond to the photon energies used for laser-induced nanostructuring.
Figure 2 and
Figure 3 reveal that the proposed algorithm performed well for most of the cases, especially to reproduce the imaginary part behaviour. The discrepancies appear for the real part of Au permittivity at 300 K and 10,000 K as the global-optimization method is less well-suited to reproduce strong local oscillations. This is also related to the fact that density functional theory calculations using the LDA potential cannot properly reproduce bound electrons and thus are not accurate for Au. However, it is interesting to note that at a higher electron temperature, the Au permittivity becomes smoother, and the Drude-Lorentz fitting model proves to be more efficient. The same statement is true for most of the metals excited at high
. For higher electron temperatures, the Fermi-Dirac distribution of electrons becomes less abrupt around the Fermi level
, spreading the allowed optical transitions on a larger energy domain. This softens the peak effects of the density of states (DOS), leading to a smoother permittivity describing the optical transitions of the smeared electrons. Electron distribution smearing intrinsically decreases the error in the calculation of optical transitions around the sharp features of the electron DOS within the
d band for transition metals.
Remarkably, the ab initio permittivity is more accurate at high electron temperatures and probably less dependent on the inherent variation of the experimental material preparation process. At a high
, the electrons occupy high-energy delocalized states, the electron density becomes more homogeneous compared to that at zero
, and the LDA potential, derived from the exchange-correlation functional of a homogeneous electron gas, becomes more accurate [
30]. As a result, although the experimental and ab initio permittivities disagree for some metals and energy ranges at 300 K (after comparing
Figure 1 and
Figure 2 and
Figure 3), that does not invalidate the calculations at higher
. The procedure to measure the optical constants is very sensitive to the experimental conditions, quality of the metal surface, and data extraction methods that may be at the origin of the disagreement at 300 K. Some other experimental data sets reported in the literature agree well with ab initio calculations [
24]. Additionally, the ab initio permittivity describes an ideal case of a pure metal and perfect illumination conditions that are hard to achieve experimentally.
Figure 1.
Drude–Lorentz fit of experimental permittivity at 300 K reported in the
Handbook on Optical Constants of Metals (Adachi, 2012) [
29]. The real
and imaginary
parts of the permittivity are shown. An electron–phonon equilibrium is considered.
Figure 1.
Drude–Lorentz fit of experimental permittivity at 300 K reported in the
Handbook on Optical Constants of Metals (Adachi, 2012) [
29]. The real
and imaginary
parts of the permittivity are shown. An electron–phonon equilibrium is considered.
Figure 2.
Drude–Lorentz fit of permittivity calculated using ab initio methods for Cr, W, and Ti. The real and imaginary parts of permittivity are shown at different electron temperatures corresponding to different levels of electron–phonon nonequilibrium.
Figure 2.
Drude–Lorentz fit of permittivity calculated using ab initio methods for Cr, W, and Ti. The real and imaginary parts of permittivity are shown at different electron temperatures corresponding to different levels of electron–phonon nonequilibrium.
Figure 3.
Drude–Lorentz fit of permittivity calculated using ab initio methods for Fe, Au, and Ni. The real and imaginary parts of permittivity are shown at different electron temperatures corresponding to different levels of the electron–phonon nonequilibrium.
Figure 3.
Drude–Lorentz fit of permittivity calculated using ab initio methods for Fe, Au, and Ni. The real and imaginary parts of permittivity are shown at different electron temperatures corresponding to different levels of the electron–phonon nonequilibrium.
Table 3.
Drude-Lorentz model fitting coefficients for the permittivity calculated using ab initio methods for Cr, W, and Ti.
Table 3.
Drude-Lorentz model fitting coefficients for the permittivity calculated using ab initio methods for Cr, W, and Ti.
| Coefficient | Cr | W | Ti |
|---|
| 300 K | 10,000 K | 25,000 K | 300 K | 10,000 K | 25,000 K | 300 K | 10,000 K | 25,000 K |
|---|
| 0.132 | 0.168 | 0.133 | 0.156 | 0.249 | 0.185 | 0.115 | 0.241 | 0.199 |
| 0.110 | 0.154 | 0.138 | 0.120 | 0.137 | 0.107 | 0.177 | 0.417 | 0.447 |
| 0.567 | 0.312 | 0.388 | 0.015 | 0.052 | 0.131 | 0.694 | 0.783 | 0.377 |
| 2.382 | 2.855 | 4.571 | 0.171 | 0.840 | 1.040 | 1.932 | 2.592 | 2.188 |
| 1.935 | 1.411 | 1.578 | 0.686 | 0.652 | 0.325 | 0.936 | 1.751 | 1.823 |
| 0.002 | 0.569 | 0.204 | 0.003 | 0.008 | 0.015 | 0.306 | 0.374 | 0.274 |
| 0.100 | 2.437 | 2.105 | 0.100 | 0.333 | 0.579 | 1.363 | 1.760 | 1.685 |
| 2.608 | 3.302 | 3.446 | 1.114 | 2.412 | 2.343 | 2.062 | 3.024 | 3.126 |
| 0.471 | 0.038 | 0.121 | 2.038 | 1.837 | 1.860 | 0.602 | 0.140 | 0.189 |
| 1.640 | 0.396 | 1.342 | 6.709 | 7.195 | 9.992 | 1.626 | 0.878 | 1.666 |
| 3.241 | 3.423 | 3.976 | 3.668 | 3.933 | 4.813 | 3.689 | 4.223 | 4.690 |
| 0.128 | 0.153 | 0.495 | 0.376 | 0.348 | 0.175 | 0.370 | 0.419 | 0.544 |
| 0.001 | 0.001 | 1.512 | 0.961 | 1.023 | 0.952 | 0.001 | 0.212 | 0.458 |
| 6.773 | 7.187 | 9.614 | 5.141 | 5.209 | 5.347 | 6.882 | 7.275 | 8.058 |
| 0.299 | 0.521 | 0.320 | 0.587 | 0.301 | 0.588 | 0.878 | 0.436 | 0.691 |
Table 4.
Drude-Lorentz model fitting coefficients for the permittivity calculated using ab initio methods for Fe, Au and Ni.
Table 4.
Drude-Lorentz model fitting coefficients for the permittivity calculated using ab initio methods for Fe, Au and Ni.
| Coefficient | Fe | Au | Ni |
|---|
| 300 K | 15,000 K | 30,000 K | 300 K | 10,000 K | 25,000 K | 300 K | 10,000 K | 25,000 K |
|---|
| 0.234 | 0.134 | 0.110 | 0.099 | 0.184 | 0.235 | 0.165 | 0.149 | 0.191 |
| 0.254 | 0.141 | 0.148 | 0.060 | 0.146 | 0.150 | 0.069 | 0.077 | 0.100 |
| 0.639 | 0.664 | 0.088 | 0.045 | 0.006 | 0.753 | 0.499 | 0.037 | 0.762 |
| 1.895 | 4.302 | 1.167 | 0.461 | 0.618 | 7.133 | 2.009 | 0.707 | 8.704 |
| 1.280 | 1.325 | 0.770 | 2.530 | 2.457 | 2.069 | 0.628 | 0.638 | 2.754 |
| 0.280 | 0.211 | 0.020 | 0.152 | 0.651 | 0.013 | 0.103 | 0.278 | 0.001 |
| 0.904 | 1.399 | 0.587 | 1.263 | 6.075 | 0.491 | 1.820 | 2.648 | 0.037 |
| 2.512 | 2.821 | 1.401 | 3.545 | 3.738 | 3.207 | 2.797 | 1.967 | 4.502 |
| 0.026 | 0.013 | 0.414 | 0.146 | 0.001 | 0.006 | 0.180 | 0.046 | 0.030 |
| 0.754 | 0.006 | 2.677 | 2.107 | 0.072 | 0.300 | 0.532 | 0.404 | 0.513 |
| 6.131 | 6.352 | 2.970 | 5.261 | 4.016 | 6.251 | 5.277 | 5.250 | 5.021 |
| 0.721 | 1.570 | 1.107 | 0.512 | 0.368 | 0.657 | 0.313 | 0.686 | 0.255 |
| 2.311 | 7.397 | 8.961 | 1.493 | 1.194 | 2.158 | 1.458 | 7.070 | 8.779 |
| 7.492 | 12.540 | 10.025 | 7.431 | 7.470 | 10.654 | 7.297 | 6.737 | 7.250 |
| 1.621 | 0.688 | 0.405 | 3.486 | 4.930 | 0.146 | 0.392 | 0.919 | 0.565 |
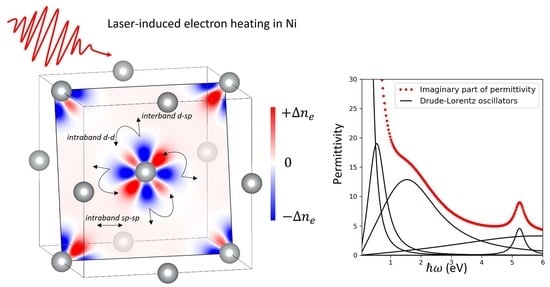
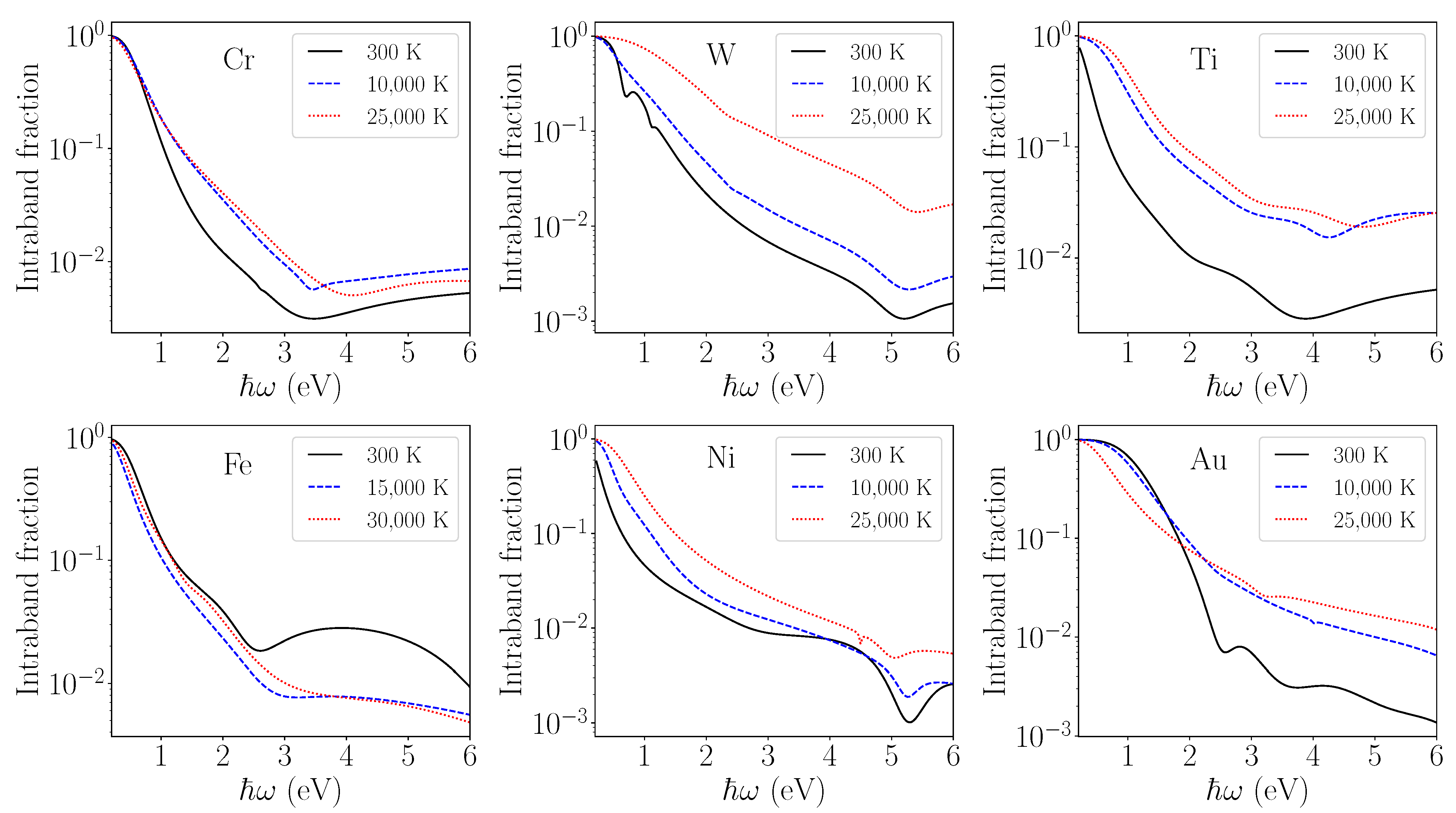
To understand other effects of a high electron temperature on permittivity, we analyzed the intraband and interband contributions separately. The contribution of intraband transitions to the permittivity generally increases with electron temperature, as shown in
Figure 4. The high electron temperature also influences the relative strength of the interband transitions and shifts the interband transition energy, as can be seen from the evolution of the optimization parameters
with temperatures from
Table 3 and
Table 4. This is also seen from
Figure 5, where the contributions to the imaginary part of permittivity from different oscillators (order 0 to 4) are plotted separately. Different terms can thus be easily identified. The intraband term (the oscillator of 0th order) is high in the infrared range below 1 eV. While the maximum is at zero photon energy limit, the intraband contribution decreases quickly at higher photon energies. The interband terms have a Lorentzian shape with different heights, widths, and locations. These features are modeled by
,
, and
values with higher order corresponding to a higher central frequency
. For some cases, the frequency of the fourth-order oscillator is higher than the chosen range and thus does not appear on the graph. Note that for some cases,
reaches the set constraint of 0.001 and thus does not provide any physical meaning. It happens, however, either for the fourth oscillator or for other insignificant oscillators (such as a tiny second oscillator for Cr). Additionally, the first-order oscillator is sometimes similar in behavior to the zero-order oscillator, as obtained by the optimization procedure (for example, for W, at 25,000 K) with both zero and first orders exhibiting a continuous decrease with
, which is characteristic of an intraband trend. In this case, the intraband contribution was considered as a sum of the zero- and first-order oscillators.
It is generally believed that for a strong laser excitation, equivalent to a high electron temperature in our case, the interband oscillators change their relative strength
(becoming stronger or weaker) and width
, whereas the central frequency
remains constant [
16]. We can see, however, in
Figure 5 that the position of the oscillator maxima changes too. For example, the third-order oscillator of Ti shifts from about 3.7 eV at 300 K to 4.7 eV at 25,000 K. Such shifts reflect the changes of the DOS of the transition metals at high excitation levels [
5] and are discussed in more detail in the following section.
4. Discussion
The optical properties of transition metals are largely defined by the features of their electronic structure. Contrary to simple metals, such as Al, Mg, or Zn, transition metals are characterized by the presence of
d-bands that translate into a high-density
d-block in the DOS [
5]. The
d-block represents highly localized electron states filled with bound-like electrons. The metallic elements used in this study are representative to capture the electronic and optical characteristics for a broad variety of transition metals. During the laser excitation, the electrons thermalize and redistribute almost instantly (on the femtosecond time scale) within the DOS [
4,
31]. The
d-block is affected by laser-induced Fermi smearing of electrons extending the energy range available for optical transitions and thus strongly impacting the permittivity. Particularly, the position of the
d-block relative to the Fermi level defines the strength of the interband component of the permittivity as a function of the photon energy. The
d-block also serves as a reservoir for populating upper free-like states, and its electronic filling degree thus defines the direct transitions within and from the
d-states. The other part of the DOS corresponds to spatially and energetically delocalized
-states and mainly defines the intraband component.
In Au, the
d-block is completely filled and separated from the Fermi level by about 2 eV. This favors significant interband transitions from the
d-band to the
-band in the visible range for photon energy higher than 2 eV with intra-
-band transitions prevailing below 2 eV. This is almost the case for Ni as well, with the Fermi level being close to the top of the
d-block and intraband transitions dominating in the infrared range. For Cr, W, Ti, and Fe, the
d-states are partially filled, fostering interband contribution for a wide range of photon energies with the intraband transitions inside the
d-band at low photon energies. The estimated relative contribution of the intraband process to the total optical transitions is presented in
Figure 4. As expected, for photons in the infrared and near-infrared region at
eV, intraband transitions are largely dominant.
For higher photon energies
eV, interband transitions dominate over all spectra for all metals, although at a higher electron temperature, the intraband role in the optical response is strengthened. For Au and Ni, a high electron temperature leads to the loss of the localized
d-electrons and the increase in the density of delocalized
-electrons that results in the shift of the
d-block towards low energies. For Cr, Fe, Ti, and W, the effect of electronic heating induces an inverse electron flow, and the
d-block shifts towards higher energies. The modification of the DOS manifested mainly by the shift of the
d-block impacts the interband contribution of the permittivity with a large effect related to the shift of the oscillator positions (
Figure 5). The Fermi–Dirac smearing at a high electron temperature significantly populates
-states while decreasing the
d-band filling, leading to the increase in the intraband fraction, as shown in
Figure 4. The opposite behavior of Fe is remarkable as due to the DOS modification, the
d-block becomes completely filled upon electron heating, fostering interband transitions under a strong nonequilibrium. The loss or gain of electronic localization and its effect on the DOS can be graphically visualized in our previous paper [
24].
The above observations underline the need to consider oscillator shifts in a nonequilibrium Drude-Lorentz model, as featured in
Figure 5. It disentangles the contributions from different oscillators to the permittivity as a function of the excitation level. The general trend that emerges is that relative contributions of the highest-order oscillators flatten towards being almost negligible at a high
. Moreover, the central frequency of the first oscillator shifts toward low energy, indicating a progressive interband-to-intraband conversion. A linear extrapolation of the fitting coefficients is possible for “hot” temperatures covering the experimental range of 5000–50,000 K (according to our tests). In this case, the Drude-Lorentz model could become an effective tool to describe the optical response of photoexcited transition metals.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




