1. Introduction
Diamonds are third-generation semiconductor materials that are often called ultimate semiconductors. Due to their excellent mechanical, chemical, and electronic properties [
1], such as high fracture strength, corrosion resistance, high thermal stability, high breakdown field, and high carrier mobility [
2] diamonds have attracted considerable attention in many fields. Doping different impurities in diamond to get the required performance is always a research hotspot, for example the Group-4 vacancy color centers in diamond can be used for quantum communication and sensor application [
3]. Wide band gap (5.5 eV) makes diamond an ideal electronic material [
1]. By doping donor and acceptor atoms in diamonds, the band gap can be adjusted to obtain p-type or n-type diamonds. P-type diamonds have already been successfully produced by doping with boron atoms and is considered as a mature topic [
4]. However, the fabrication of high-quality n-type diamonds remains challenging [
5].
Doped impurities in n-type diamonds primarily include lithium (Li), sodium (Na), oxygen (O), nitrogen (N), sulfur (S), and phosphorus (P). They are present in diamonds either as interstitial atoms (such as Li and Na) or substitutional atoms (such as O, N, S, and P) [
6]. N, S, and P attracted considerable attention in previous studies [
7]. Unlike N, which is proven to be a deep donor with an activation energy of 1.7 eV [
8] and S with an activation energy of more than 1 eV and low doping efficiency as demonstrated by theoretical research [
9], P with a relatively lower donor level (0.6 eV) is currently considered the best substitutional donor [
4]. Theoretical and experimental studies prove that the diamond growth rate appreciably increases as a function of doping phosphorus and thereby reduce the production costs [
10]. Koizumi et al. [
11] show that n-type diamonds can be successfully produced by doping phosphorus using microwave-enhanced plasma chemical vapor deposition (MPCVD).
However, it is difficult to obtain practical n-type diamonds by doping P atoms. Satio et al. [
12] proved that in P-doped (111) diamond surfaces, the carrier concentration and Hall mobility at 500 K are only 3.8 × 10
8 cm
3 and 38 cm
2/V, respectively, and the Hall mobility in (001) surfaces is even lower. Koizumi et al. report that the Hall mobility of activated carriers is very low (28 cm
2/V at 500 K) and decrease as the temperature decrease (vanishing below 300 K). They suggest that unsatisfactory electrical properties may be due to crystalline imperfections caused by the incorporation of a large amount of P atoms, because crystalline perfection has a significant influence on the electrical properties [
11,
13]. The Hall mobility vary with the temperature, also suggesting that crystal defects are dominant factors in electron scattering [
14]. Through positron lifetime measurement, Dannefaer et al. [
15] show that CVD diamond films usually contain a high concentration of vacancies (>50 ppm). The vacancy-related defects in diamonds are commonly considered as deep acceptors that act as strong donor compensation centers. Ideal P-doped diamonds should contain a large amount of P atoms and also be free of vacancies [
16]. Therefore, a thorough understanding of the formation and structure of vacancy-related complexes in P-doped diamonds are important and worth studying. H atoms are also the reason for carrier compensation [
17]. A large amount of H exists during CVD diamond growth [
11,
13], and more than 10
19 cm
−3 of hydrogen is detected on (001) surfaces [
18]. It is also necessary to study H-related complexes in detail. Theoretically, the doping mechanisms can be summarized by two methods, doping on the surface and in the crystal [
19]. In previous studies, PV
xH
y complexes in bulk diamonds were carefully investigated [
16,
20], but PV
xH
y complexes on growing surfaces still lack detailed research.
Because formation energy of PV and PVH complex is relatively low in bulk diamond [
20], indicating that they are more likely to form than other PV
xH
y complexes, for simplicity, only PV and PVH complexes are considered in this work. The purpose of this study is to investigate the formation of PV and PVH complexes and their influence on the surface structure within diamond (001) and (111) surfaces based on first-principles calculations, since (001) and (111) surfaces are the most commonly used surfaces for doped diamonds. Recently there are many researches in P-doping experiments on (110) surface [
21], but as this orientation is difficult to obtain due to its unstable nature during growth, we do not study it in this work. By analyzing the band structure of bulk diamond with PV and PVH complexes, Yan C. X. et al. find that PV complex is an acceptor center providing hole carrier in diamond and PVH complex introduces a deep acceptor level in band gap [
20]. We also investigate doping single P atom and forming single V in diamond surfaces for comparison to reveal the possibility of the emergence of PV and PVH complexes during P atom doping and structural deformation due to vacancies and hydrogen atoms.
2. Materials and Methods
In this study, the Cambridge Sequential Total Energy Package (CASTEP) [
22] based on density function theory (DFT) is used for all of the calculations. An ultrasoft pseudopotential [
23] plane wave approach is adopted to describe the electrons. The Perdew-Burke-Ernzerhof (PBE) and general gradient approximation (GGA) [
24] are used for the exchange–correlation functional. The cutoff energy is 400 eV and the 2 × 2 × 1
k points for both (001) and (111) surfaces are used to sample the Brillouin zone according to the Monkhorst-Pack method [
25]. Spin-polarization is taken into consideration [
26]. The convergence criterion of the inter-atomic forces is set to be 0.03 eV/ Å, and the energy of self-consistent calculation is 1.0 × 10
−5 eV/atom. The cutoff energy and
k points are obtained by careful testing and are proven to be adequate for all of the calculations in this work; higher values of these parameters shows a difference within only 5% in calculation of formation energy.
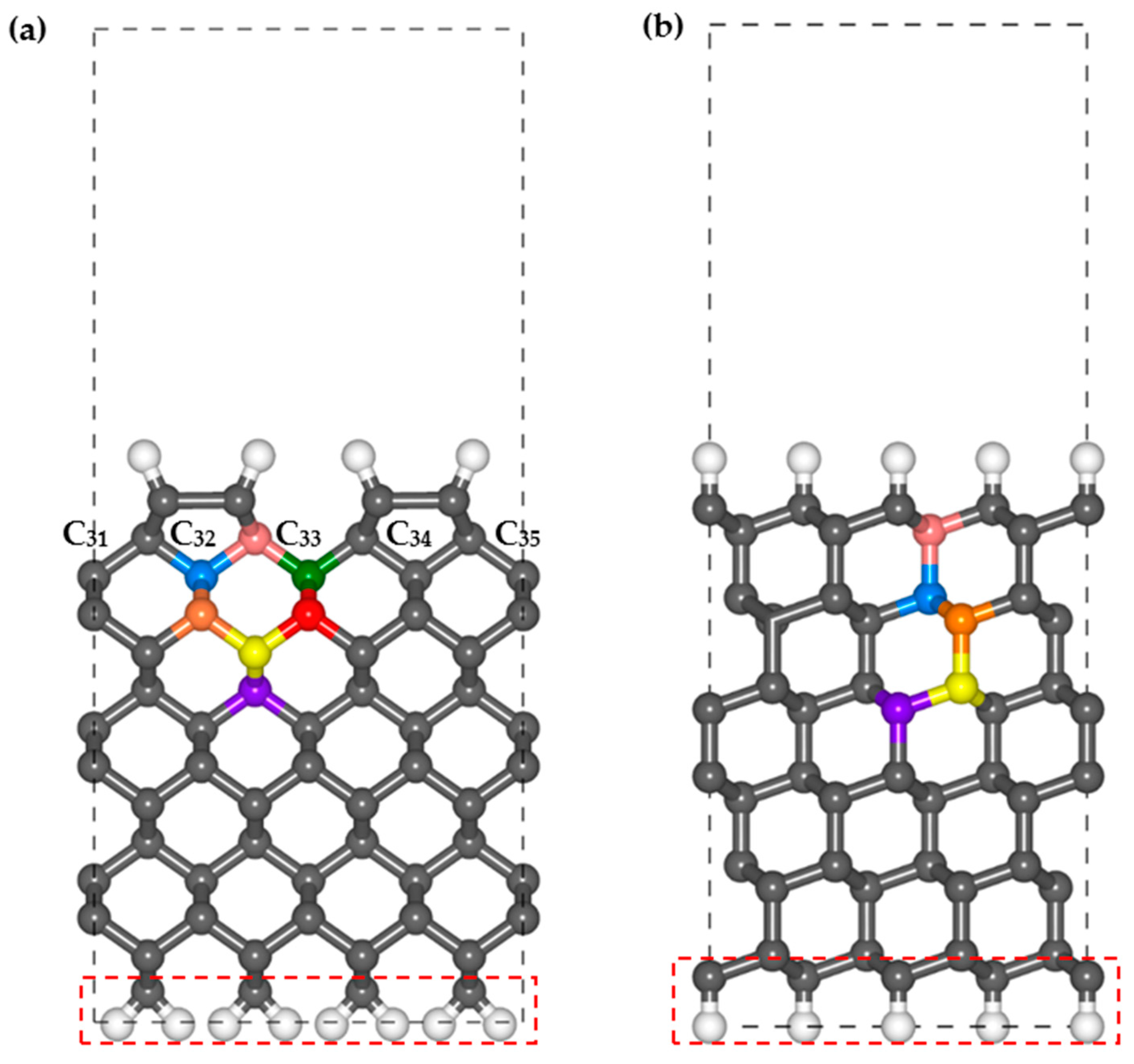
The H-terminated diamond 2 × 1 reconstructed (001) and (111) surfaces are used in this study as they are the most common CVD-growth diamond thin films. A vacuum distance of 10 Å is adopted between the periodical slabs in the
z direction to avoid interactions. The numbers of C layers on the (001) and (111) surfaces are 14 and 12, respectively, which are also obtained using the test calculations. The bottom C layer and terminated H layer are constrained to simulate continuous bulk diamonds in the lower region of the surface structure. The calculation parameters and lattice parameters are listed in
Table 1.
The formation energy is calculated using the following equation [
27,
28]:
where
n and
m are the number of vacancies and H atoms, respectively. In this study, it only investigates the situation that they are either zero or one.
and
represent the total energy of the diamond surface containing
and the pure diamond surface, respectively.
,
, and
denote the chemical potentials of P, H, and C, respectively, deduced from their hydrides in gas phase (PH
3, H
2), and bulk diamonds.
is the result of exchanges of electrons or holes with the carrier reservoirs [
28]. The following section focuses only on the
of impurities with neutral charges, so the last term is zero.
4. Conclusions
The effects of vacancy and hydrogen on the growth and morphology of n-type phosphorus-doped C (001)-2×1:H and C (111)-1×1:H diamond surfaces are theoretically studied in this investigation. The research subjects included single P atoms, PV complexes, and PVH complexes. The calculations are based on DFT under periodic boundary conditions. The properties included: (i) the formation energies of doping a single P atom, PV complex, and PVH complex, (ii) surface structural deformation caused by dopants, and (iii) bond populations of the chemical bonds around the dopants. Based on our calculations, the following conclusions are drawn from the results:
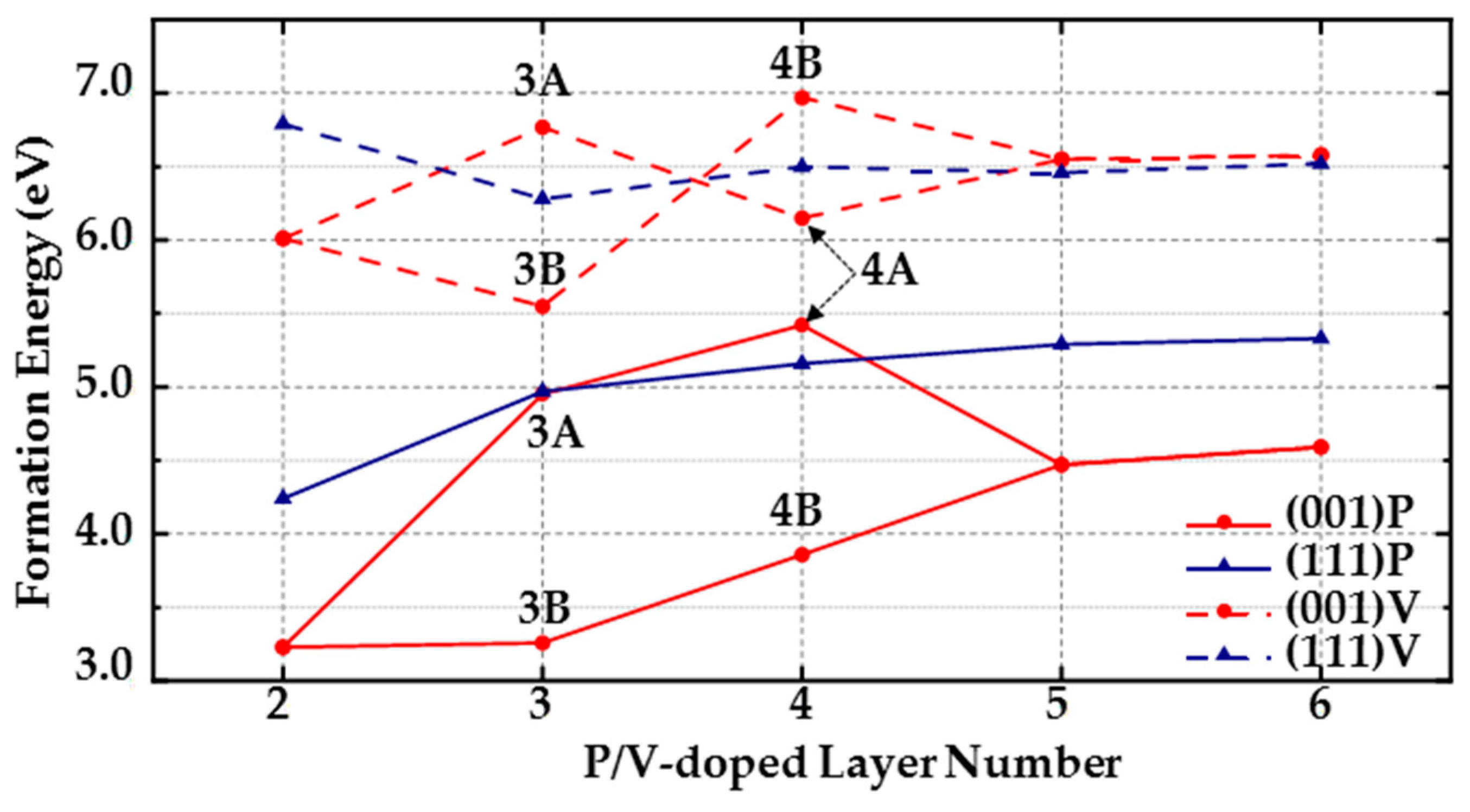
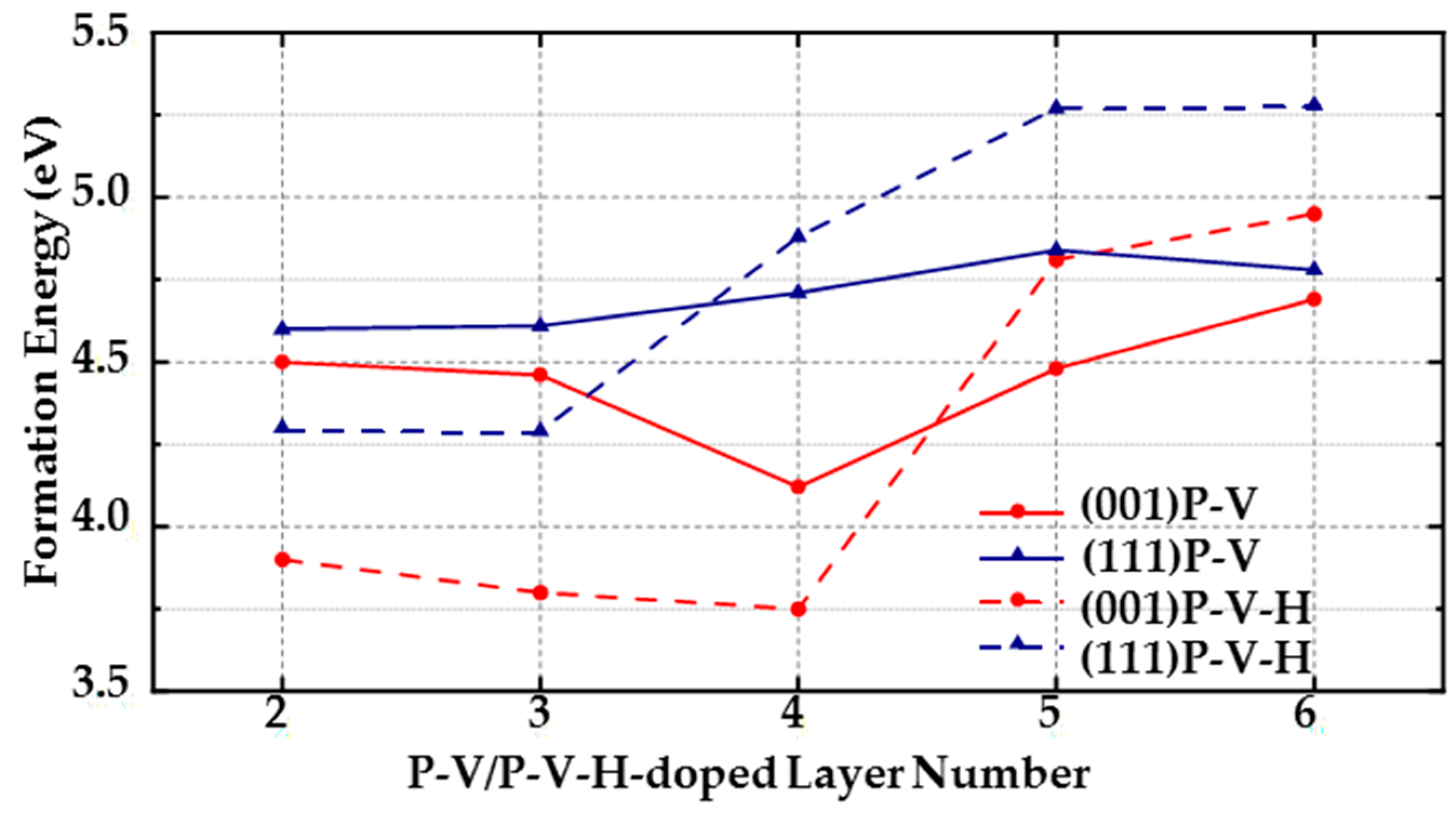
(1) For (111) diamond surfaces, the formation energy of the PV complex is lower than the formation energy of a single P atom in the third, fourth, fifth, and sixth layers. In the third and fourth layers, the formation energy of a single P atom is even higher than that of the PVH complex, indicating that when doping P atoms on (111) surfaces, the doped P atoms are likely to form PV and PVH complexes with the surrounding vacancies and H atoms. Thus, it is very important to control the number of vacancies and H atoms around doped P atoms during MPCVD fabrication of (111) diamond surfaces.
(2) The formation energy of a single P atom is generally the lowest in (001) surfaces in the second to sixth layers, which means that compared with a single P atom, PV and PVH complexes are less likely to form on (001) surfaces (but PV and PVH complexes form more easily than on (111) surfaces because the lowest formation energies of PV and PVH complexes in each layer of (001) surfaces are all lower than those of (111) surface. The lowest formation energy of doping a P atom in each layer of (001) surface is at least 0.74 eV lower than that of (111) surface). However, in the third and fourth layers, the formation energy of PVH complexes is close to or lower than the formation energy of doping a single P atom. For example, the lowest formation energy of PVH complexes in the fourth layer is 3.75 eV, and the lowest formation energy of doping a P atom in the fourth layer is 3.86 eV. Thus, decreasing the amount of hydrogen in the formation process of (001) diamonds can effectively reduce defects.
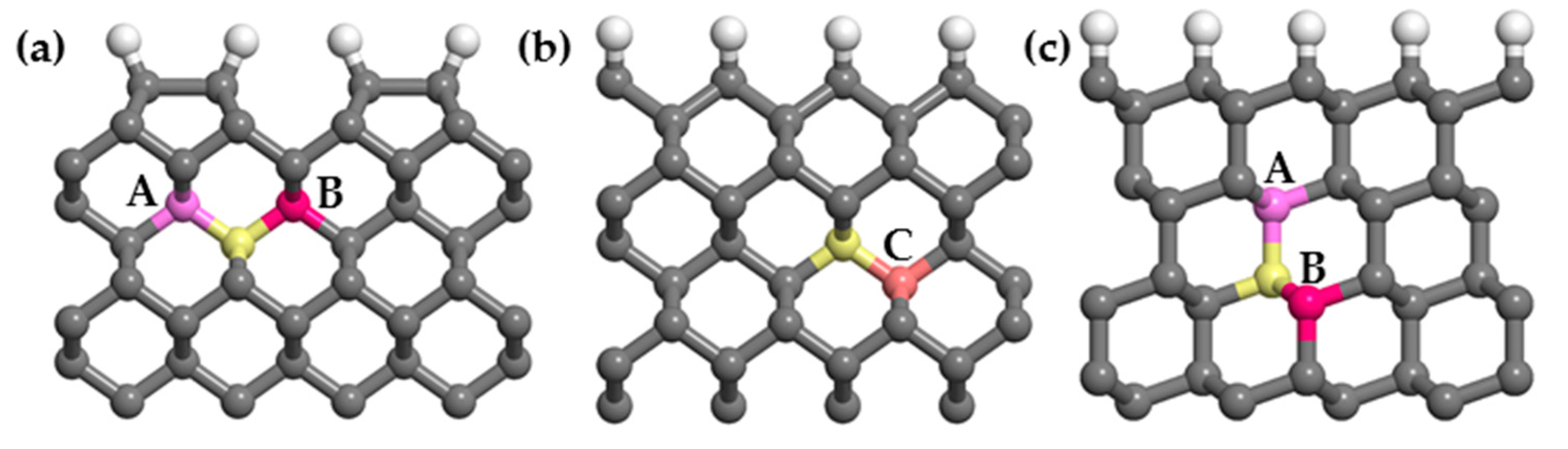
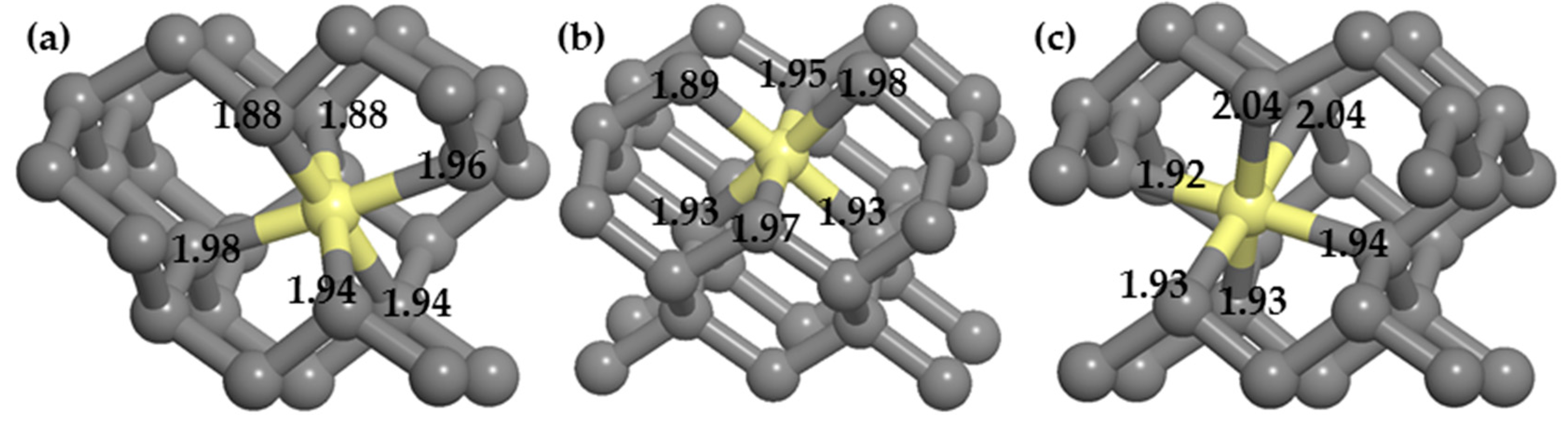
(3) Dopants cause surface structural deformation, and the formation energy is partly related to the extent of deformation. When the structural deformation is insufficient, distortions caused by doping are limited and eventually increase the formation energy, similar to doping a single P atom. When the structural deformation is excessive, more energy is required to reconstruct the surrounding structures, similar to the PV and PVH complexes.
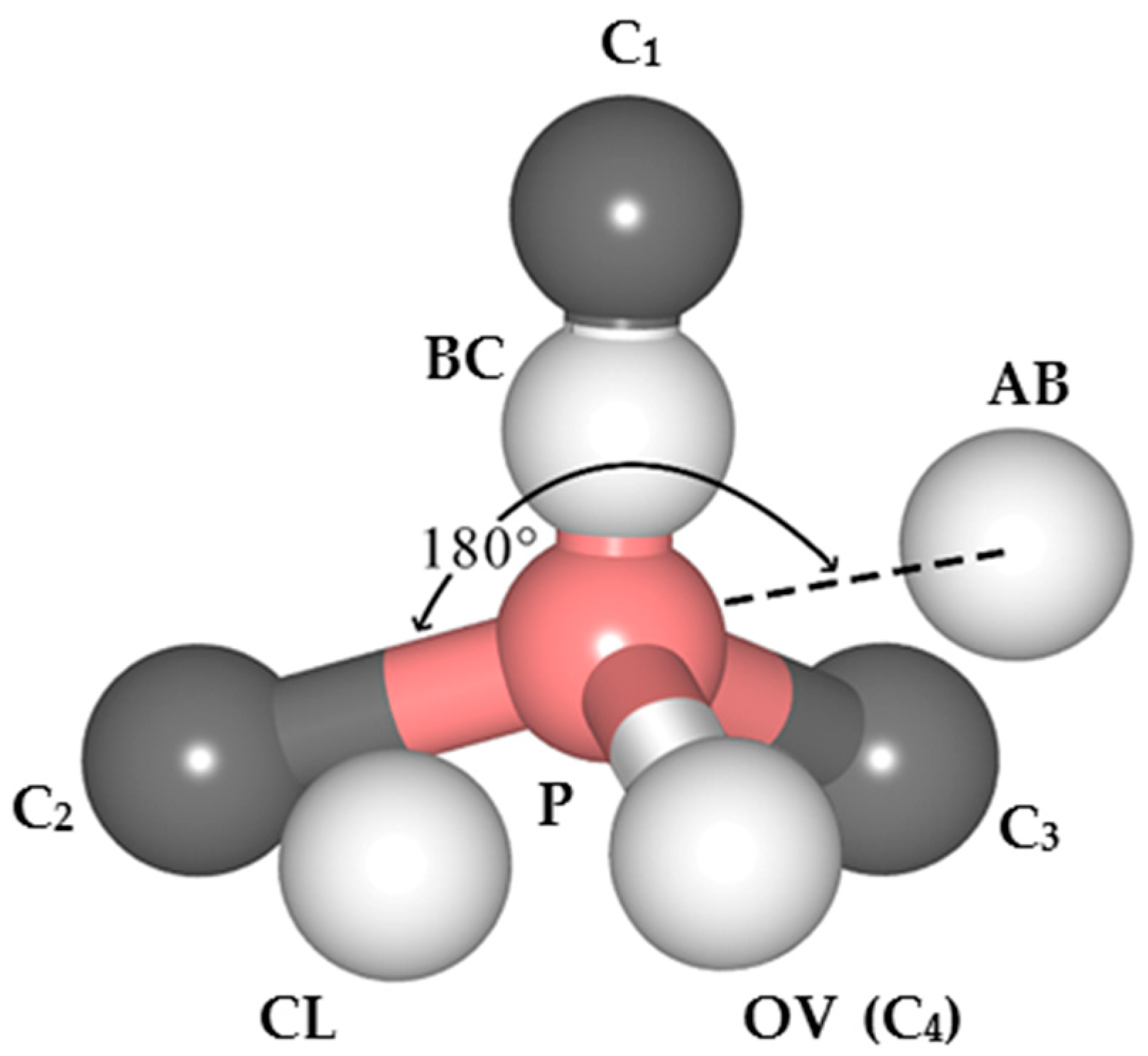
(4) Around the PVH complex, the P atom does not bond with the H atom, but instead bonds with the five nearby carbon atoms. Since phosphorus has only five valence electrons, compared with the six C–P bonds around PV complexes, the P–C bond populations around PVH complexes are higher, indicating that the P–C bonds around PVH complexes are stronger than those around PV complexes and P–C complexes are more stable than PV complexes.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}