
Fluorescence Correlation Spectroscopy Combined with Multiphoton Laser Scanning Microscopy—A Practical Guideline


Abstract
:1. Background
2. Concepts and Theory
2.1. Principles of FCS
- The concentration correlation is independent on absolute time t and will only depend on the lag-time τ.
- At short time intervals, typically orders of magnitude shorter than the diffusion time of the molecules, the value of the correlation function will approach the equilibrium state, i.e.,
- For long time intervals, the fluctuations will become uncorrelated, i.e.,
- The measurements are carried out with very dilute, close to ideal solutions so that statistics of solute molecules are independent.
- Concentration fluctuations of different species must be uncorrelated.
- The mean-square fluctuation in a unit volume follows Poisson statistics. This means that the variance of the fluctuation will increase as mean number of recorded signal increases.
2.2. Experimental Requirements FCS-MPM
2.3. Autocorrelation Using TCSPC Data
3. Experimental Section
3.1. Optical Setup
3.2. Chemicals
3.3. FCS Data Analysis
4. Results and Validation
4.1. Effect of Numerical Aperture (NA) on FCS Measurements
4.2. Effect of Time Binning on FCS Measurements
4.3. Effect of Concentration on FCS Measurements
4.4. Effect of Excitation Power on FCS Measurements
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef]
- Sheppard, C. Scanning Confocal Microscopy. Encycl. Opt. Eng. 2003, 2525–2544. [Google Scholar] [CrossRef]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996, 59, 427–471. [Google Scholar] [CrossRef]
- Kirejev, V.; Guldbrand, S.; Borglin, J.; Simonsson, C.; Ericson, M.B. Multiphoton microscopy—A powerful tool in skin research and topical drug delivery science. J. Drug Deliv. Sci. Technol. 2012, 22, 250–259. [Google Scholar] [CrossRef]
- Konig, K. Multiphoton microscopy in life sciences. J. Microsc. 2000, 200, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Schwille, P.; Haupts, U.; Maiti, S.; Webb, W.W. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys J. 1999, 77, 2251–2265. [Google Scholar] [CrossRef] [Green Version]
- Haustein, E.; Schwille, P. Annual Review of Biophysics and Biomolecular Structure (ANNU REV BIOPH BIOM). Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 151–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwille, P. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem. Biophys. 2001, 34, 383–408. [Google Scholar] [CrossRef]
- Schwille, P.; Heinze, K.G. Two-Photon Fluorescence Cross-Correlation Spectroscopy. ChemPhysChem 2001, 2, 269–272. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar] [CrossRef]
- Magde, D.; Elson, E.; Webb, W.W. Thermodynamic Fluctuations in a Reacting System—Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett. 1972, 29, 705–708. [Google Scholar] [CrossRef]
- Elson, E.L.; Magde, D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974, 13, 1–27. [Google Scholar] [CrossRef]
- Rigler, R.; Mets, Ü.; Widengren, J.; Kask, P. Fluorescence correlation spectroscopy with high count rate and low background: Analysis of translational diffusion. Eur. Biophys. J. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Heikal, A.A.; Hess, S.T.; Baird, G.S.; Tsien, R.Y.; Webb, W.W. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: Coral red (dsRed) and yellow (Citrine). Proc. Natl. Acad. Sci. USA 2000, 97, 11996–12001. [Google Scholar] [CrossRef] [Green Version]
- Hess, S.T.; Webb, W.W. Focal Volume Optics and Experimental Artifacts in Confocal Fluorescence Correlation Spectroscopy. Biophys. J. 2002, 83, 2300–2317. [Google Scholar] [CrossRef] [Green Version]
- Wachsmuth, M.; Waldeck, W.; Langowski, J. Anomalous diffusion of fluorescent probes inside living cell investigated by spatially-resolved fluorescence correlation spectroscopy. J. Mol. Biol. 2000, 298, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Schwille, P.; Oehlenschlager, F.; Walter, N.G. Quantitative hybridization kinetics of DNA probes to RNA in solution followed by diffusional fluorescence correlation analysis. Biochemistry 1996, 35, 10182–10193. [Google Scholar] [CrossRef]
- Widengren, J.; Mets, Ü.; Rigler, R. Fluorescence correlation spectroscopy of triplet states in solution: A theoretical and experimental study. J. Phys. Chem. 1995, 99, 13368–13379. [Google Scholar] [CrossRef]
- Schwille, P.; Korlach, J.; Webb, W.W. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry 1999, 36, 176–182. [Google Scholar] [CrossRef]
- Kim, S.A.; Heinze, K.G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963–973. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer Science+ Business Media: Berlin, Germany, 2006; pp. 1–954. [Google Scholar]
- Wang, Z.; Shah, J.V.; Chen, Z.; Sun, C.H.; Berns, M.W. Fluorescence correlation spectroscopy investigation of a GFP mutant-enhanced cyan fluorescent protein and its tubulin fusion in living cells with twophoton excitation. J. Biomed. Opt. 2004, 9, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Guldbrand, S.; Kirejev, V.; Simonsson, C.; Goksör, M.; Smedh, M.; Ericson, M.B. Two-photon fluorescence correlation spectroscopy as a tool for measuring molecular diffusion within human skin. Eur. J. Pharm. Biopharm. 2013, 84, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Benham, G.S. Chapter 6—Practical Aspects of Objective Lens Selection for Confocal and Multiphoton Digital Imaging Techniques. In Methods in Cell Biology; Matsumoto, B., Ed.; Academic Press: Cambridge, MA, USA, 2002; Volume 70, pp. 245–299. [Google Scholar]
- Török, P.; Kao, F. Optical Imaging and Microscopy; Springer: Berlin/Heidelberg, Germany, 2007; Volume 87. [Google Scholar] [CrossRef]
- Young, M.D.; Field, J.J.; Sheetz, K.E.; Bartels, R.A.; Squier, J. A pragmatic guide to multiphoton microscope design. Adv. Opt. Photonics 2015, 7, 276–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrean, A.; Mansvelder, H.D. Optimal lens design and use in laser-scanning microscopy. Biomed. Opt. Express 2014, 5, 1588–1609. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Bergmann, A.; Haustein, E.; Petrasek, Z.; Schwille, P.; Biskup, C.; Kelbauskas, L.; Benndorf, K.; Klöcker, N.; Anhut, T.; et al. Fluorescence lifetime images and correlation spectra obtained by multidimensional time-correlated single photon counting. Microsc. Res. Tech. 2006, 69, 186–195. [Google Scholar] [CrossRef]
- Felekyan, S.; Kühnemuth, R.; Kudryavtsev, V.; Sandhagen, C.; Becker, W.; Seidel, C.A.M. Full correlation from picoseconds to seconds by time-resolved and time-correlated single photon detection. Rev. Sci. Instrum. 2005, 76, 1–14. [Google Scholar] [CrossRef]
- Becker, W. Advanced time-correlated single photon counting techniques. In Springer Series in Chemical Physics; Springer Science & Business Media: Berlin, Germany, 2005; Volume 81, pp. 1–387. [Google Scholar]
- Phillips, D.; Drake, R.C. Time Correlated Single-Photon Counting (Tcspc) Using Laser Excitation. Instrum. Sci. Technol. 1985, 14, 267–292. [Google Scholar] [CrossRef]
- Böhmer, M.; Wahl, M.; Rahn, H.-J.; Erdmann, R.; Enderlein, J. Time-resolved fluorescence correlation spectroscopy. Chem. Phys. Lett. 2002, 353, 439–445. [Google Scholar] [CrossRef]
- Becker, W. The bh TCSPC Handbook, 8th ed.; Becker & Hickel: Berlin, Germany, 2019; p. 968. [Google Scholar]
- Stock, R.S.; Ray, W.H. Interpretation of photon correlation spectroscopy data: A comparison of analysis methods. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 1393–1447. [Google Scholar] [CrossRef]
- James, J.; Kantere, D.; Enger, J.; Siarov, J.; Wennberg, A.-M.; Ericson, M. Report on fluorescence lifetime imaging using multiphoton laser scanning microscopy targeting sentinel lymph node diagnostics. J. Biomed. Opt. 2020, 25, 071204. [Google Scholar] [CrossRef]
- Laurence, T.A.; Fore, S.; Huser, T. Fast, flexible algorithm for calculating photon correlations. Opt. Lett. 2006, 31, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Petrášek, Z.; Schwille, P. Photobleaching in Two-Photon Scanning Fluorescence Correlation Spectroscopy. ChemPhysChem 2008, 9, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Berland, K.; Shen, G. Excitation saturation in two-photon fluorescence correlation spectroscopy. Appl. Opt. 2003, 42, 5566–5576. [Google Scholar] [CrossRef] [PubMed]
- Fick, A.V. On liquid diffusion. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1855, 10, 30–39. [Google Scholar] [CrossRef]










{kind=link}
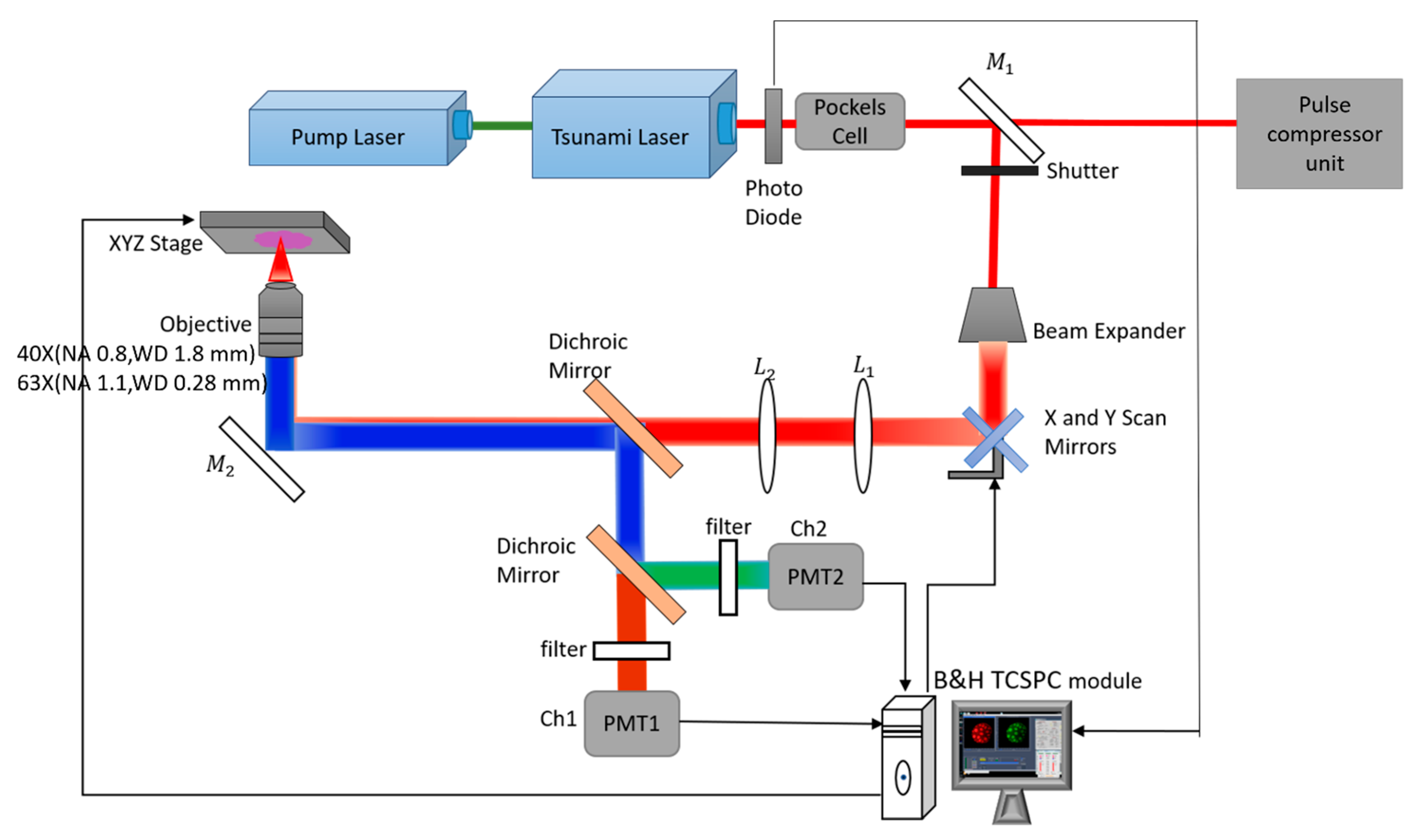{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
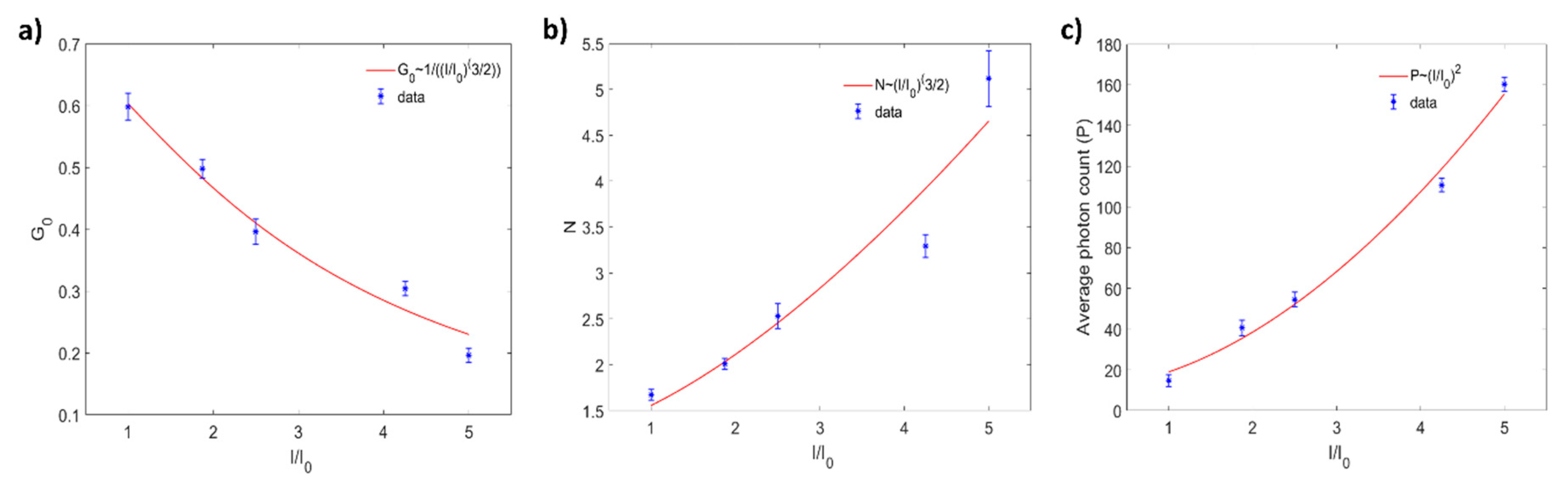{kind=link}
{kind=link}
| Parameters | Conditions | Observations and Recommendations |
|---|---|---|
| Objective lens | High NA objective lenses (NA > 1) are preferred for FCS; however, for imaging MPM the long working distances normally restricts NA. |
|
| Concentration range | The concentration should preferably be in nM range for FCS, but for imaging the concentration range can vary by several orders of magnitudes. | Concentration ranges needs to be validated prior to measurements:
|
| Excitation power | In MPM-FCS the excitation power will determine the excitation and detection volume. |
|
| Time binning | TCSPC raw data has a sparse binary data format, and time binning is required in order to perform autocorrelation. | The optimal range of time binning should be validated for the specific experiments.
|
| Autocorrelation algorithm | Multi-tau and extended data fitting models have been proposed in the literature. Here it is shown that linear-tau and simple model fitting is preferred for the data analysis. |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, J.; Enger, J.; Ericson, M.B. Fluorescence Correlation Spectroscopy Combined with Multiphoton Laser Scanning Microscopy—A Practical Guideline. Appl. Sci. 2021, 11, 2122. https://doi.org/10.3390/app11052122
James J, Enger J, Ericson MB. Fluorescence Correlation Spectroscopy Combined with Multiphoton Laser Scanning Microscopy—A Practical Guideline. Applied Sciences. 2021; 11(5):2122. https://doi.org/10.3390/app11052122
Chicago/Turabian StyleJames, Jeemol, Jonas Enger, and Marica B. Ericson. 2021. "Fluorescence Correlation Spectroscopy Combined with Multiphoton Laser Scanning Microscopy—A Practical Guideline" Applied Sciences 11, no. 5: 2122. https://doi.org/10.3390/app11052122
APA StyleJames, J., Enger, J., & Ericson, M. B. (2021). Fluorescence Correlation Spectroscopy Combined with Multiphoton Laser Scanning Microscopy—A Practical Guideline. Applied Sciences, 11(5), 2122. https://doi.org/10.3390/app11052122