3.1. Preparation of the Electrodes and Characterization
Photocathodes based on nanocrystalline NiO were prepared according to procedures adapted from the literature [
19]. Two different methodologies were employed, leading to NiO electrodes of type A and type B. For NiO electrodes of type A, a NiO paste made of pre-formed NiO nanoparticles was employed and deposited by spin coating onto FTO electrodes, while for NiO electrodes of type B, a precursor solution based on NiCl
2 was used, which was deposited onto FTO by doctor blading (see
Section 2.2). In both cases, calcination at 550 °C following a programmed ramping procedure was performed to attain the desired NiO electrodes. Film thickness was measured using a profilometer and found to be 1.1 and 0.6 μm for electrodes of types A and B, respectively (
Figure S3). The morphology of the NiO films was then characterized by SEM and AFM analyses. Representative SEM micrographs and the corresponding AFM images are shown in
Figure 2.
For both electrode types, the SEM analysis shows a homogeneous distribution of the nanocrystalline NiO support onto the FTO substrate. The surface is, however, more porous and rougher for NiO electrodes of type A (
Figure 2A) with respect to NiO electrodes of type B, for which a flatter structure is clearly apparent (
Figure 2B). Both AFM images (
Figure 2C,D) and the corresponding section analyses (
Figure S4) further confirm these observations. Increased surface roughness was indeed measured for electrodes of type A (Rq = 48.4 nm, Ra = 40.9 nm) when compared to electrodes of type B (Rq = 13.7 nm, Ra = 10.6 nm). These data agree with the higher porosity, as measured by gas adsorption isotherms, and the smaller crystallite size, as measured by X-ray diffraction (XRD) analysis, expected for NiO electrodes of type A rather than those of type B [
19]. Finally, the AFM analysis (
Figure 2C,D and
Figure S4) suggests improved connectivity among NiO nanoparticles in the case of electrodes of type A.
The NiO films were then sensitized by overnight soaking into a 0.5 mM solution of 4-(bis-{4-[5-(2,2-dicyano-vinyl)-thiophene-2-yl]-phenyl}-amino)-benzoic acid (
P1) in acetonitrile. Two exemplars of the resulting electrodes are reported in
Figure 1C,D. Since electrodes of type A are not sufficiently transparent to enable reliable measurement of the absorption spectrum using spectrophotometry in transmission mode, determination of the dye loading was attained through a desorption procedure performed by treating a sensitized electrode with tetrabutylammonium hydroxide (TBAOH) in methanol followed by registration of the absorption spectrum (see
Supplementary Materials, Figure S5). Then, considering the molar extinction coefficient of the
P1 dye in acetonitrile at the absorption maximum (ε = 58,000 M
−1cm
−1) [
17] and assuming similar ε values for the protonated and deprotonated species, the amount of dye was quantified using the Lambert–Beer equation. A surface concentration of 11.7 (±1.7) nmol∙cm
−2 was estimated for sensitized NiO electrodes of type A. On the other hand, determination of the dye loading for sensitized electrodes of type B was possible by direct measurement of the absorption spectrum (
Figure S6). Using the Lambert–Beer equation for thin films (Equation (1)) and considering the maximum absorption and the extinction coefficient [
17], a surface loading of 6.9 (±0.3) nmol∙cm
−2 was calculated. This result was further confirmed by desorption measurements with TBAOH in methanol (surface concentration of 6.7 (±0.9) nmol∙cm
−2,
Figure S7). The larger dye loading observed for NiO electrodes of type A can be attributed to the higher porosity of the NiO film as well as to the larger film thickness, both leading to an increased surface area for attachment of the
P1 dye.
Deposition of heterogeneous Pt was performed on sensitized NiO electrodes immersed in the platinizing solution based on H
2PtCl
6 as the metal precursor [
22] using simple and fast procedures upon application of a potential bias or a combination of both potential and light biases. Five different methodologies were considered. In Methods 1 and 2, a stationary potential of −0.2 V vs. SCE was applied, either coupled or not to light irradiation, and different time intervals were considered. Under these conditions, Pt deposition implies the reduction of Pt(IV) to Pt(0), most likely occurring through the transient formation of a Pt(II) species [
24]. The typical current vs. time responses obtained using Methods 1 and 2 are reported in the
Supplementary Materials (Figure S8). Both feature an increase in cathodic current, ascribable to nucleation and progressive growth of Pt onto the sensitized NiO surface, until reaching a plateau associated with deposition conditions limited by diffusion of Pt(IV) to the electrode surface [
25]. Interestingly, the rates to achieve such plateau currents are different for the two methods, suggesting that the nucleation and growth of Pt islands onto sensitized NiO might differ to some extent when the photoexcited dye can mediate the reduction of Pt(IV) ions as in Method 1 (i.e., under irradiation conditions after hole injection into the NiO valence band) or not (i.e., under dark conditions, Method 2). Following the suggestions in the literature [
26], we also employed potentiodynamic conditions for Pt attachment onto our sensitized NiO electrodes (Methods 3–5). Three cycles of cyclic voltammetry spanning the potential range between +0.3 and −0.2 V vs. SCE at a scan rate of 0.1 V·s
−1 were considered, either coupled or not to light irradiation (see
Section 2.2). We envisioned that the application of the negative potential enabling the reduction of Pt(IV) will promote the nucleation of Pt metal onto the surface, while the subsequent anodic scan will renew the Pt(IV) electrolyte solution close to the electrode, thus potentially favoring a cleaner deposition upon the subsequent cathodic scan. The current vs. voltage traces recorded during the platinization process of sensitized NiO electrodes of type A are reported in the
Supplementary Materials (Figures S9–S11). An increase in cathodic current was observed in the presence of the light bias, possibly due to the active participation of the
P1 dye in the deposition event [
14]. Interestingly, a noticeable current enhancement was also observed at negative potentials, regardless of the method used, in the second and third scans, which can be attributed to progressive, favorable growth of Pt islands following kinetically limited nucleation at the first cathodic scan [
26].
Combined SEM/EDS and AFM analyses were employed to characterize exemplars of sensitized and platinized NiO electrodes.
Figure 3 depicts the data for two electrodes of type A obtained via photoelectrodeposition, viz. Method 1 with a deposition time of 20 s and Method 5, whereas the results for an exemplar prepared via the dark Method 2 are reported in the
Supplementary Materials (Figure S12). For all the samples tested, the observation of characteristic S and Pt emissions in the EDS maps clearly confirms the simultaneous presence of both the
P1 dye and the Pt HEC, respectively. Furthermore, both the EDS and AFM analyses suggest that regardless of the deposition method, the distribution of the Pt metal is rather homogenous, supporting preferential formation onto the sensitized NiO surface of small, well-distributed Pt islands over large metal clusters. The failure to observe, at the current size resolution, appreciably different morphological features in the case of light vs. dark conditions possibly suggests that such differences are, if at all present, intrinsically small.
3.2. Photoelectrochemical Characterization
The NiO photoelectrodes functionalized with both
P1 dye and the Pt HEC, according to the procedures previously discussed, were tested by means of linear sweep voltammetry (LSV) and chronoamperometry (CA) under chopped light irradiation (calibrated at 1 sun) in N
2-purged 0.1 M acetate buffer at pH 4. The associated maximum photocurrent density, viz. the maximum difference between the measured current density in the presence of light subtracted by that in the absence of light, was then extracted as a figure of merit for comparison purposes (see the estimate for an exemplar in
Figure S13). The experimental conditions were fixed because at a more acidic pH, rapid discoloration of the electrode was observed, ascribed to substantial desorption of the
P1 dye from the NiO surface, while at a higher pH, the resulting photocurrents were considerably smaller than those measured here, thus preventing a reliable evaluation of the different deposition methodologies. Furthermore, since we aimed for a fast screening of the deposition methodology, the CA measurements were limited to a time window of 5 min.
Figure 4 shows the relevant LSV curves recorded for the sensitized and platinized NiO photoelectrodes of type A. As can be clearly seen, all platinization procedures, regardless of their nature, yielded active photocathodes with measured photocurrent densities featuring onset potentials between 0.4 and 0.7 V vs. SCE and values well above those of sensitized NiO without Pt. This experimental evidence can be taken as a clear indication that all functionalized electrodes can effectively promote electron transfer processes to the electrolyte mediated by the Pt metal on the surface. In particular, the observed improved photocurrents can be ascribed to the occurrence of photoinduced electron transfer processes involving hole injection from the excited state of the
P1 dye (*
P1) into the valence band (VB) of NiO, followed by electron transfer from the reduced form of the dye (
P1−) to the Pt HEC that mediates proton reduction to dihydrogen (
Figure 1A). More importantly, the measured photocurrent densities appreciably depend on the procedure employed for the attachment of the catalytic metal.
For electrodes prepared using Method 1 (hereafter named NiO-A|
P1|Pt-
1; see
Figure 4A for two relevant exemplars and
Figure S14 for the whole series), the maximum photocurrent densities achieved were also affected by the deposition time: −15 µA·cm
−2 at 10 s, −25 µA·cm
−2 at 20 s, −15 µA·cm
−2 at 30 s, −18 µA·cm
−2 at 50 s, and −13 µA·cm
−2 at 100 s. Interestingly, the largest values were attained at short to intermediate deposition times (i.e., 20 s), most likely resulting as the best balance in terms of catalyst loading. A similar effect was already observed in our previous study using CdTe/CdS quantum dots as sensitizers [
16]. A large catalyst loading for NiO-A|
P1|Pt-
1 electrodes at longer deposition times (see, e.g., 100 s in
Figure 4A) was further confirmed by the noticeable increase in cathodic dark current, likely ascribable to direct electrocatalysis by Pt. The trend in photocurrent densities for the different deposition times was also maintained when the electrodes were probed by CA at −0.2 V vs. SCE (
Figure S15), although the steady-state photocurrent values measured were lower. Interestingly, the samples prepared at longer photoelectrodeposition times (>30 s) showed rapidly decreasing photocurrents attributable to charge recombination processes, most likely determined by the presence of large amounts of Pt HEC on the electrode surface.
Method 2, which differs from Method 1 in the absence of the light bias, was also employed for the preparation of NiO photocathodes (hereafter named NiO-A|
P1|Pt-
2). Previous studies that employed Method 2 as a platinization procedure usually considered long deposition times (>300 s) [
27,
28]. Attempts at preparing NiO-A|
P1|Pt-
2 electrodes with such deposition times were, however, unsuccessful, as negligible photocurrent densities, close to those of the blank sample, as well as considerable dark currents, ascribed to direct electrocatalysis by Pt, were measured. Shorter deposition times, similar to those employed in Method 1, were thus considered. These electrodes systematically performed worse than those obtained with Method 1, leading to maximum photocurrent densities of −10 µA·cm
−2 at 10 s and −12 µA·cm
−2 at 20 s as measured by LSV under chopped irradiation (
Figure 4B). This suggests that the coupling of both light and electrochemical potential is important to favor effective Pt deposition onto sensitized NiO electrodes. A similar trend was observed also in the CA measurements (
Figure S16 of the Supplementary Materials).
The best results obtained using both Methods 1 and 2 at short deposition times clearly evidence the necessity of having a small amount of catalytic Pt metal on the sensitized NiO surface. Large amounts of catalyst may indeed play the role of recombination centers, thus decreasing the possibility of profitably converting photogenerated electrons into molecular hydrogen [
25,
29]. In this respect, we envisioned that the application of an inherently fast procedure such as cyclic voltammetry might well comply with this important requirement. As a matter of fact, the potentiodynamic deposition methods (Methods 3–5) turned out to be effective for the platinization of sensitized NiO electrodes, with performances that depended on the presence and duration of the light bias.
Figure 4C shows the LSV traces obtained under chopped irradiation of electrodes prepared according to Methods 3–5 (hereafter named NiO-A|
P1|Pt-
3, NiO-A|
P1|Pt-
4, and NiO-A|
P1|Pt-
5, respectively), whereas in
Figure S17, the corresponding CA traces recorded at −0.2 V vs. SCE are reported. Maximum photocurrent densities in the order of −29, −27, and −40 µA·cm
−2 were measured for the NiO-A|
P1|Pt-
3, NiO-A|
P1|Pt-
4, and NiO-A|
P1|Pt-
5 electrodes, respectively. According to these data, Method 5, which combines a CV scan and a short illumination, provides the most active photocathodes. Although the SEM/EDS analysis suggests a similar distribution of the Pt HEC onto the sensitized NiO surface, the beneficial effect of including light irradiation under both potentiostatic and potentiodynamic conditions is clearly apparent. The resulting improvement most likely stems from subtle structural arrangements as well as NiO/dye/catalyst interactions that might differ when the
P1 dye takes an active part (as in Methods 1, 3, and 5) or does not (as in Methods 2 and 4) in the reduction of the Pt(IV) electrolyte and the nucleation of Pt.
The NiO electrodes obtained via the best deposition procedures among the potentiostatic and potentiodynamic methods (viz. NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5) were further characterized to gain additional insight into the photoelectrochemical performances. We first checked if the observed photocurrent enhancements were assignable to hydrogen generation by determining the Faradaic efficiency (FE) using a generator–collector method adapted from the literature [
20]. Hydrogen was produced with FEs of 38 (±2)% and 60 (±5)% for NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5, respectively (
Figures S18 and S19), which substantially surmount the negligible FE measured in the absence of the Pt HEC (3 (±2)%,
Figure S20). These data clearly confirm that the improvement in terms of photocurrent densities can be unambiguously assigned to profitable charge transfer to the aqueous electrolyte involving proton reduction to dihydrogen mediated by the Pt HEC, as previously inferred. The photoaction spectra of both NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5 were then determined and compared with that of the sensitized NiO electrode without Pt HEC (
Figure 4D). For all samples tested, the photoaction spectrum closely resembled the absorption spectrum of the
P1 dye [
17], thus confirming that in all cases, the photoelectrochemical response is triggered by light absorption by the molecular chromophore. More interestingly, higher efficiencies were measured in the platinized electrodes, with maximum incident photon-to-current conversion efficiency (IPCE) values of 0.15% and 0.18% for NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5, respectively. Taking Equation (2) and considering a light-harvesting efficiency (LHE) of ~0.8 at the absorption maximum—as possibly estimated according to Equations (1) and (3) considering the surface concentration of
P1 dye previously calculated (~11.7 nmol·cm
−2) and the molar extinction coefficient at the absorption maximum (ε = 58,000 M
−1cm
−1) [
17]—these data correspond to absorbed photon-to-current conversion efficiency (APCE) values of 0.187% and 0.225% for NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5, respectively. According to Equation (2) and the almost unitary yield of hole injection (Φ
inj) by the
P1 dye into NiO (~97%) [
30], charge collection efficiencies (η
cc) of 0.19% and 0.23% can be estimated for NiO-A|
P1|Pt-
1 at 20 s and NiO-A|
P1|Pt-
5, respectively.
The same platinization procedures used for electrodes of type A were employed for the attachment of the Pt HEC onto NiO electrodes of type B sensitized with the
P1 dye (hereafter named NiO-B|
P1|Pt-
n, where n = 1–5) in order to evaluate the potential general applicability of our deposition methods.
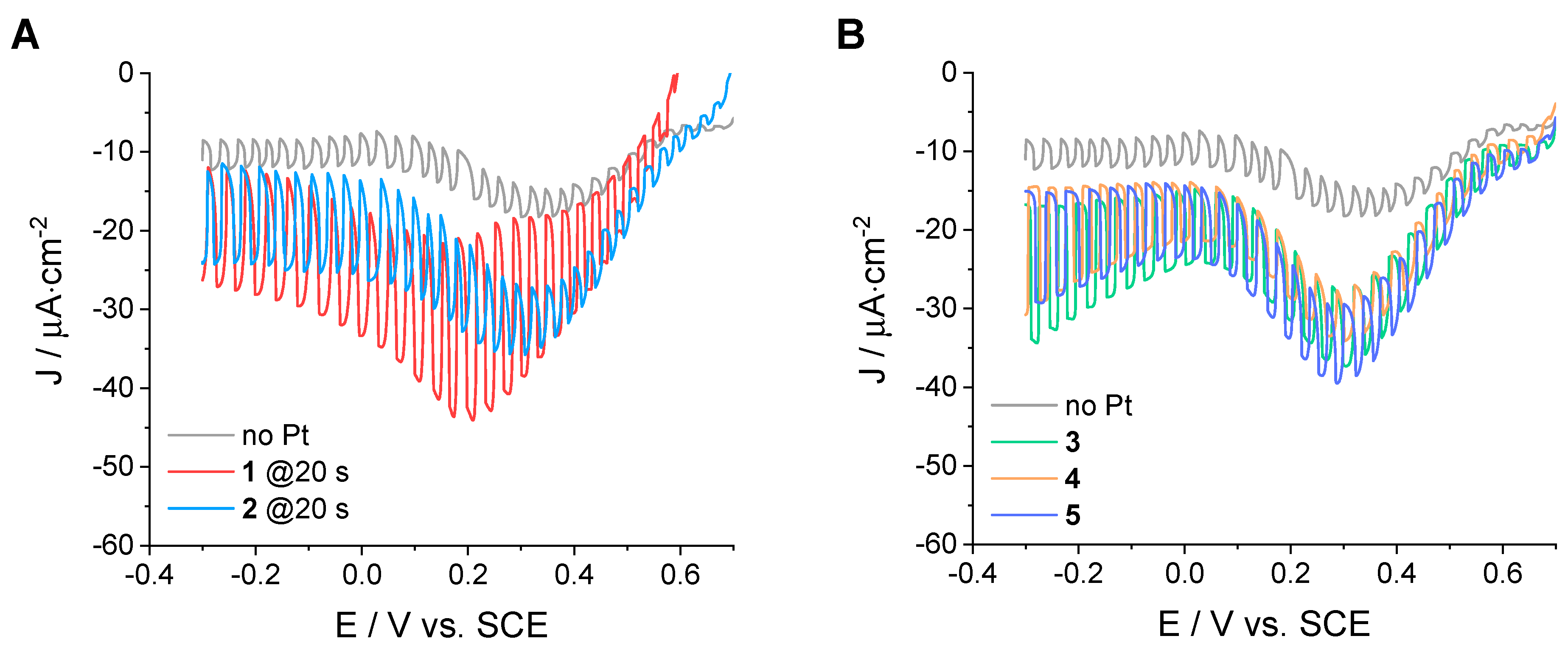
Figure 5 shows the relevant LSV curves obtained with such electrodes under chopped irradiation in 0.1 M acetate buffer at pH 4 (the corresponding CA experiments at an applied bias of −0.2 V vs. SCE are reported in the
Supplementary Materials, Figures S21–S23). Similar to type A NiO electrodes, different deposition times were considered for Methods 1 and 2 (
Figures S24 and S25), and the best performances were obtained at deposition times of 20 s (
Figure 5A). The main evidence from the comparison of these data can be summarized as follows: (i) akin to NiO electrodes of type A, the attachment of Pt onto the sensitized NiO surface provides catalytic sites for proton reduction to dihydrogen, leading to enhanced photocurrent densities relative to those observed with the blank sample without metal on top; (ii) the measured photocurrent densities are overall lower than those observed with NiO electrodes of type A, which can be attributed to the reduced dye loading and corresponding light-harvesting ability as well as to the different morphology of the NiO film (mainly size and connectivity of the NiO nanoparticles), which might influence the efficiency of charge recombination phenomena [
31]; (iii) within this limitation, potentiostatic methods (Methods 1 and 2) perform slightly better than the CV-based ones (Methods 3–5); (iv) the potentiostatic Method 1, which combines both potential bias and light, provides more active photocathodes (maximum photocurrent densities of −22 µA·cm
−2 for the NiO-B|
P1|Pt-
1 sample at 20 s deposition time) than the analogue Method 2 without applied illumination (maximum photocurrent densities of −13 µA·cm
−2 for the NiO-B|
P1|Pt-
2 sample at 20 s deposition time; see
Figure 5A); (v) as to the potentiodynamic procedures (Methods 3–5), improved performances are attained with the NiO-B|
P1|Pt-
3 electrode (maximum photocurrent densities of −18 µA·cm
−2) with respect to both NiO-B|
P1|Pt-
4 and NiO-B|
P1|Pt-
5 (maximum photocurrent densities of −14 and −15 µA·cm
−2, respectively); (vi) similar to what was observed for NiO electrodes of type A, the importance of merging both potential and light is clearly evident for both potentiostatic and potentiodynamic methods, suggesting that the investigated deposition protocols could be well suited for future studies aimed at photoelectrochemical hydrogen generation using different sensitized NiO substrates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




