1. Introduction
High-temperature solid oxide fuel cells (SOFCs) are made entirely of solid materials. As a solid electrolyte, they use metal oxides which, unlike carbonate electrolytes, do not corrode the anode and cathode. The metal-oxide material used as a solid electrolyte should be as dense and thin as possible in order to reduce the ohmic resistance. On the one hand, the high operating temperature of SOFCs has the advantage that these devices can run on a variety of fuels, including natural gas. Moreover, their operation does not require catalysts based on noble metals (for example, Pt). When reusing waste heat in SOFC, its efficiency can be brought up to 70–75%. However, the high operating temperatures of SOFCs create certain difficulties. Manufacturing SOFCs requires high thermal stability of materials, which increases the cost of the system. There is also difficulty in connecting different components in SOFC at high temperatures. The use of proton conductors exhibits to achieve the higher ionic conductivity at lower temperatures (~500–600 °C). The possibility of creating a solid ionic conductor with a lower operating temperature based on LaScO
3 perovskite was studied. At high pressure, a solid solution with a perovskite type structure was formed [
1]. For the synthesized material of the composition (Li
0.4Ce
0.15La
0.67)ScO
3, an ionic conductivity of 1.1 × 10
−3 S·cm
−1 at 623 K was achieved. A number of works have been devoted to the development of protonic electrolytes [
2,
3,
4,
5,
6,
7]. Each of them is dedicated to a particular system of materials or manufacturing technology. Sufficiently high conductivity (~10
−3 S·cm
−1) was observed for LaNbO
4 doped with calcium in a humid atmosphere at 800 °C [
8]. Importantly, this result was achieved using materials that do not contain Ba or Sr, which are not suitable for fuel cells operating in a CO
2 atmosphere.
LaScO
3 perovskite is a promising proton conductor [
9,
10,
11,
12]. It exhibits a fairly high structural stability. Even at high impurity levels, LaScO
3 is characterized by orthorhombic symmetry in the range from room temperature to about 1993 K [
13]. The structural skeleton of this compound is made up of rigid ScO
6 octahedra. The formation of oxygen vacancies, which increase the proton conductivity, occurs when La is replaced by Sr [
14]. The proton conductivity (6 × 10
−3 S·cm
−1) obtained upon reaching the ratio of Sr to La x = 0.2, i.e., for the La
0.8Sr
0.2ScO
3–δ compound, still remains one of the highest [
15]. This doped perovskite is a promising proton-conducting electrolyte and can be used in proton–ceramic fuel cells. However, a targeted enhancement of proton conductivity requires a fundamental study of the mechanism of proton migration in the corresponding solid electrolyte. Among the most reliable methods for studying the mechanism of conduction performed at the atomic level are atomistic calculations in combination with neutron diffraction and nuclear magnetic resonance (NMR). The possible implementation of the proton dynamics in a crystal with a perovskite structure has not yet been fully investigated [
16].
We used an ab initio molecular dynamics approach to comprehensively investigate the proton dynamics in a defect-free perovskite LaScO3 crystal. In this new study, we investigated the influence of initial proton velocities and locations, as well as the influence of electric field and temperature on proton behavior in LaScO3 perovskite. Carrying out these studies can confirm or refute the hypothesis about the polaron character of proton movement along the perovskite crystal lattice. Doped ceramics LaScO3 is also promising material for the use as a solid electrolyte in SOFCs.
The aim of this work is to investigate the mechanism of proton movement in defect-free LaScO3 perovskite as a function of temperature, magnitude of the applied electric field, initial velocity of the proton, and its primary location.
2. Materials and Methods
All calculations were performed using the SIESTA software package. These ab initio molecular dynamics calculations are based on the density functional theory and are implemented within the plane wave model. For all considered atoms, only valence electrons were taken into account in the calculations. The calculation of the exchange–correlation functional was based on the Perdew–Burke–Ernzerhof (PBE) formalism [
17]. In our calculations, the periodic supercell consisted of 16 La atoms, 16 Sc atoms, and 48 oxygen atoms. The initial configuration of the perfect LaScO
3 system is shown in
Figure 1. The resulting parameters of the initial lattice were: a = 11.6003997 Å, b = 8.107899684 Å, c = 11.328470354 Å, α = β = γ = 90°, and the volume of the created system was 1068.87 Å
3. Projecting the plane-wave onto specific orbitals allowed us to compute local states and populations. The Monkhorst–Pack algorithm [
18] was used to generate all 10 × 10 × 1 k-points. The cutoff energy of plane waves was 400 Ry. This was found to be sufficient to achieve convergence in the context of calculating relative energies. The integration of the equals of motion in realization of ab initio molecular dynamics was carried out by the Verlet method with a time step of 1 fs.
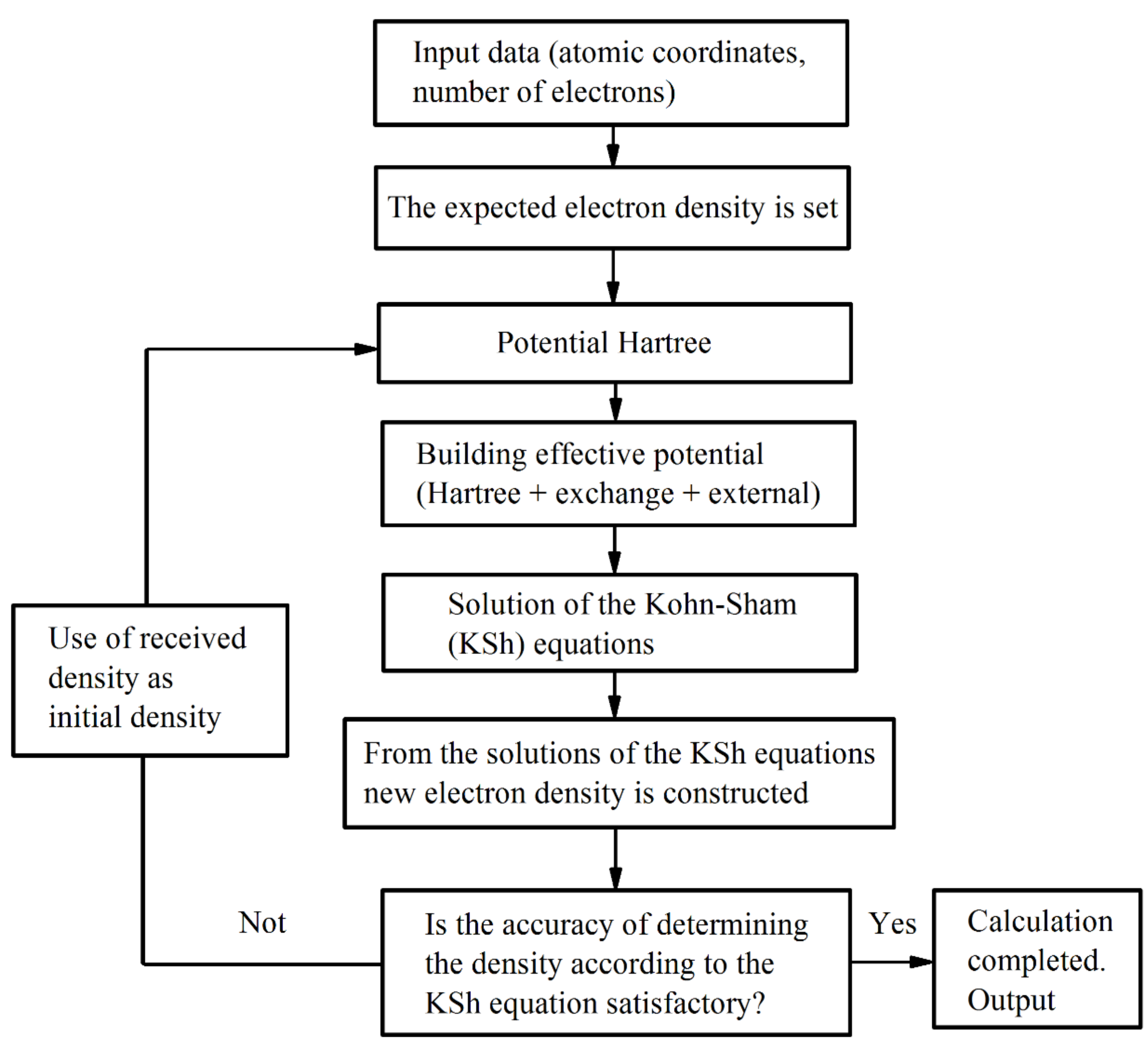
The calculations were performed according to the block diagram shown in
Figure 2. The input data are the charges of the nuclei, the number of electrons, and the coordinates of the atoms. Plane waves were used as basic functions. The calculation procedure was completed in accordance with the specified criterion for the accuracy of determining the density.
Before each ab initio molecular dynamics calculation, geometric optimization was performed. The reference value for the change in the total energy of the system during the dynamic relaxation of atoms was 0.001 eV. As a result of this preliminary preparation, a structure relaxed to a stable state was obtained, the final stress (pressure) in which was equal to 0.0007 Ry/Bohr3. Next, a proton was placed in the resulting system, which occupied different positions:
- (1)
in the Sc octahedron plane between the oxygen atoms at point 1, which has average coordinates (0.44, 0.75, 0.23),
- (2)
in the Sc octahedron plane near the oxygen atom at point 2, which has average coordinates (0.56, 0.75, 0.23),
- (3)
in the plane of finding La atoms at localization near the oxygen atom at point 3, which has average coordinates (0.33, 0.75, 0.94).
In each of these three cases of a specific initial position of the proton, five coordinates were used, differing in x and z coordinates. Accordingly, for each case (1-3), five different molecular dynamics calculations were performed. The average values of the initial coordinates of the proton (mentioned above) are also the initial coordinates of the proton for one of the runs in each series.
First-principle molecular dynamics calculations were performed for systems containing a proton. The initial velocity of the proton varied from 0 to 0.37418 × 10
5 m/s (from 0 to 0.5 Bohr/fs). An electric field acted on the proton, and the intensity vector was in the
xz plane. The directions of this vector varied, and the magnitude of the tension varied from 0 to 4.95 × 10
10 V/m (from 0 to 4.95 V/Å). Note that the maximum value of the electric field strength specified here approaches the value of the atomic electric field, which appears as a result of the interaction of atoms in perovskite oxide SrTiO
3 (4 × 10
11 near the oxygen atom and 10
12 V/m near the Ti and Sr atoms) [
19]. However, it is significantly higher than the field strength (10
3–10
4 V/m) used to perform the cycle of intercalation/deintercalation of lithium in the molecular dynamics simulation of the silicene anode functioning [
20].
Nine series of five runs each with ~1000 time steps were performed in the temperature range from 153 K to 1100 K. The last temperature value is close to the operating temperature of the solid-state fuel cells, using LaScO
3-based compounds as the electrolyte. Data on the temperature, applied electric field, initial velocity of the proton, and its primary location are presented in
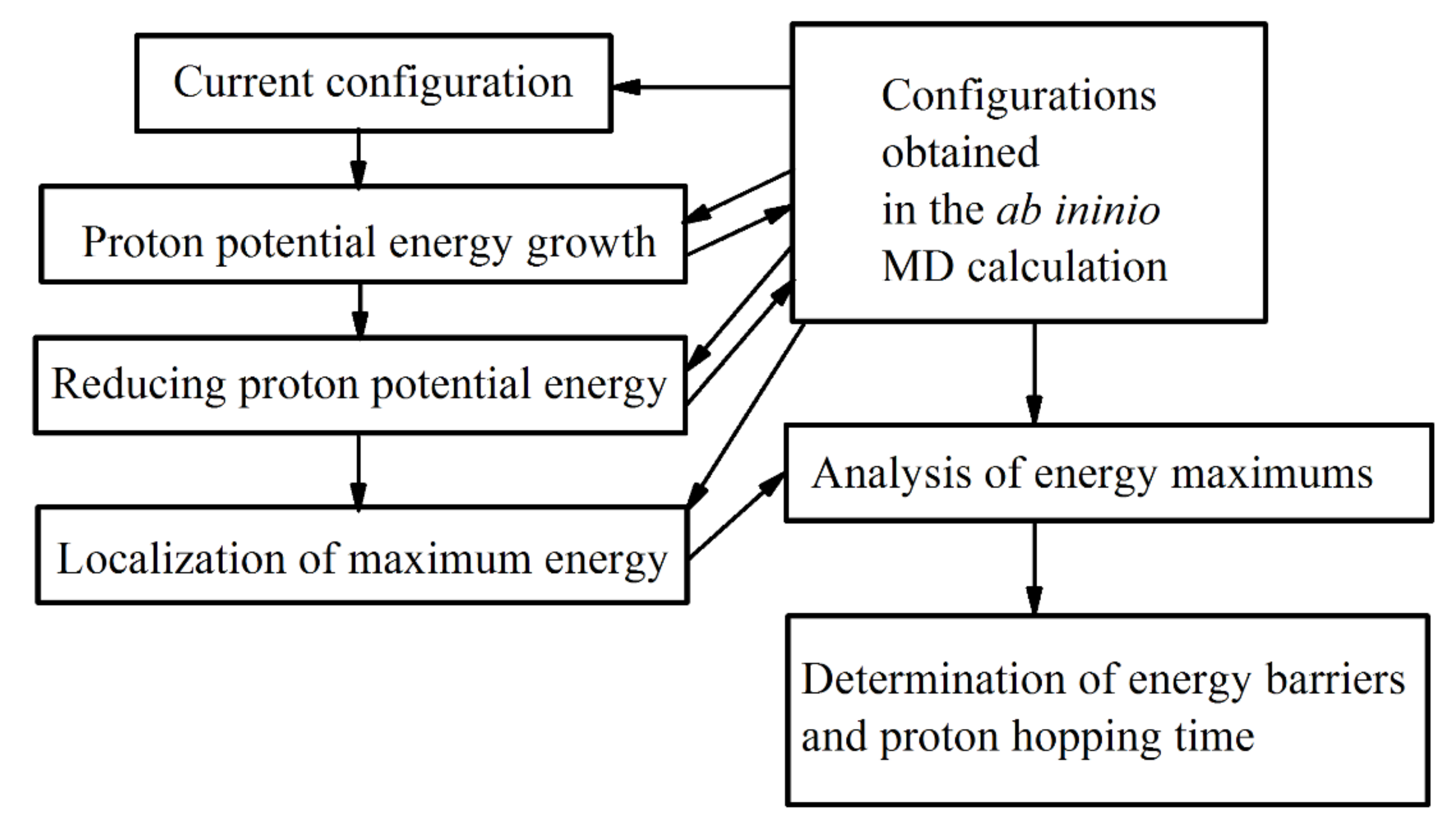
Table 1. To determine the moments of time, at which the transition of a proton from one oxygen atom to another took place, an appropriate algorithm was developed, the code of which was implemented in the Python 3.8 language. The block diagram of the program for determining the energy barrier and the time of the proton transition between oxygen atoms is shown in
Figure 3. This algorithm is based on tracking the change in the potential energy of the proton. The peaks in this dependence characterize the change in the average virtual location of the proton. The time interval between these peaks determines the time of its transition from one oxygen atom to another.
We used the method of constructing Voronoi polyhedrons to trace the mutual arrangement of the proton and the atoms of the LaScO
3 compound closest to it. Polyhedra with the proton in the center were built at each time step. The polyhedra were constructed for three separate subsystems: oxygen, scandium, and lanthanum. Thus, the geometric neighbors closest to the proton were established for O, Sc, and La for the entire time of its presence in the system. Due to the small size of the system and the short residence time of the proton in it, we do not consider the changes occurring in its complete environment, since they are small. That is, we will not present the distribution of polyhedra by the number of faces and the distribution of faces by the number of sides that characterize the rotational symmetry of the subsystems under consideration. Instead, we will consider the angular placement of the proton relative to different kinds of atoms. The angles under consideration are formed by a proton and a pair of atoms included in the immediate environment of the proton. The environment is defined using the Voronoi polyhedron. There is a proton at the vertex of the angle under consideration, the sides of the angle are formed by segments, which extreme points are the proton and one of the atoms of the subsystem under consideration. We have previously performed a similar sounding to the structure by moving a charged particle (in this case, a proton) [
21].
3. Results
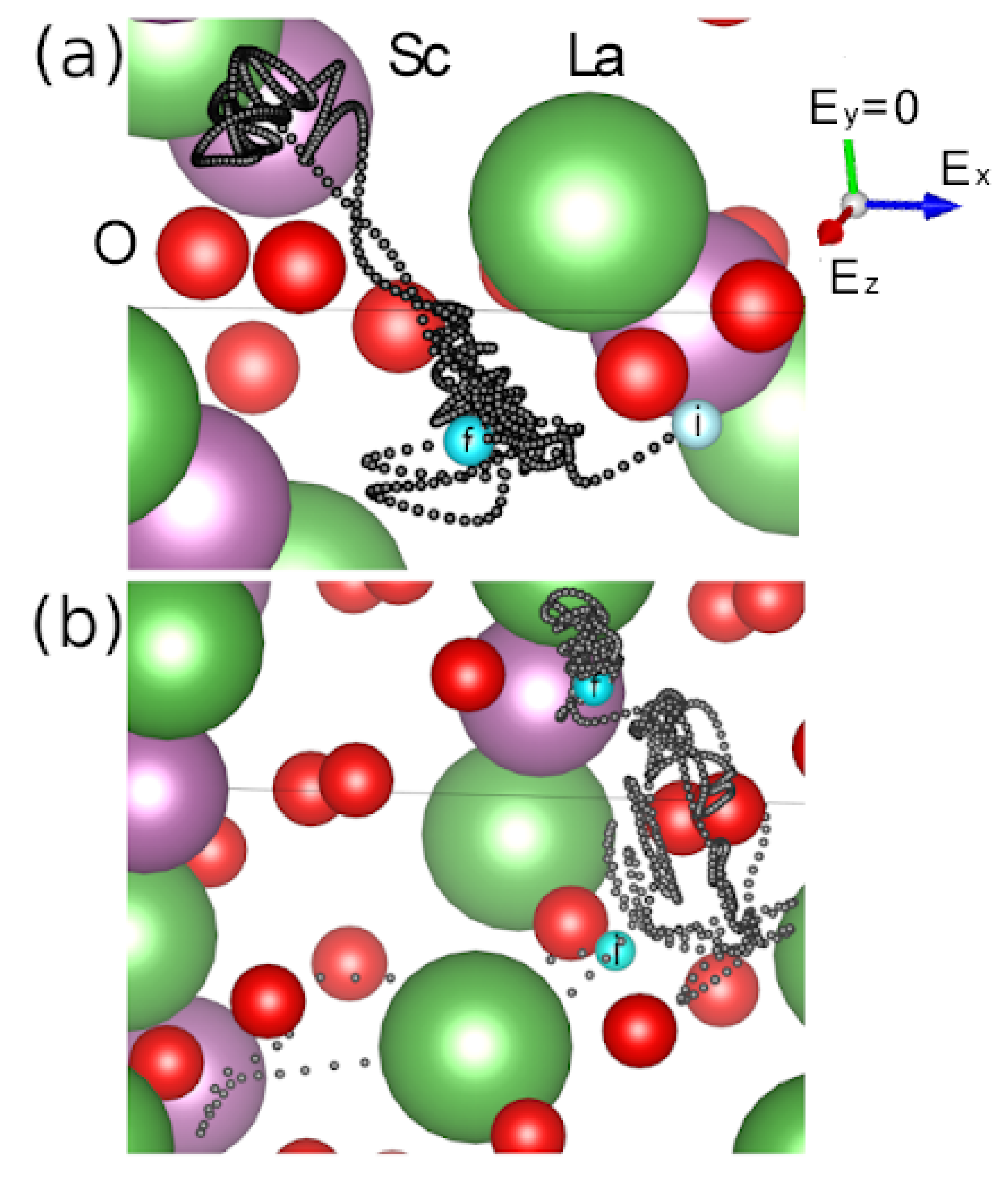
Let us trace the trajectories of a proton in cases, where its initial placement corresponds to its average initial coordinates obtained in five calculations of the same type. Horizontal projections of the configurations of the LaScO
3 crystal corresponding to the time instant of 1 ps are shown in
Figure 4.
The upper figures reflect the above system obtained in series 1, and the lower figures show a similar system resulting from the series 3. In series 1, a constant electric field with an intensity E = 0.7071 × 1010 V/m acted on the proton, the series 3 was performed in the absence of an electric field. The trajectory of a proton is enclosed in a more limited space under the effect of the electric field. It is noteworthy that a large part of the path of the proton does not pass along the direction determined by the strength of the electric field (line in the figure), but in the direction forming a certain angle with the direction of the vector E. In either case, the trajectory of the ion turns out to be rather entangled. It can get into the vicinity of another (different from the original) oxygen atom and stay there for some time. It can also approach adjacent La and Sc atoms, which are not initially adjacent to it. In addition, the proton can not only move away from its initial location, nor approach it again, moving in the direction opposite to the direction of the passed path. Thus, the electric field does not affect the choice of the preferred location for the proton in the LaScO3 crystal. Most likely, the place of its virtual location is determined by the attraction of a proton to any closest oxygen atom.
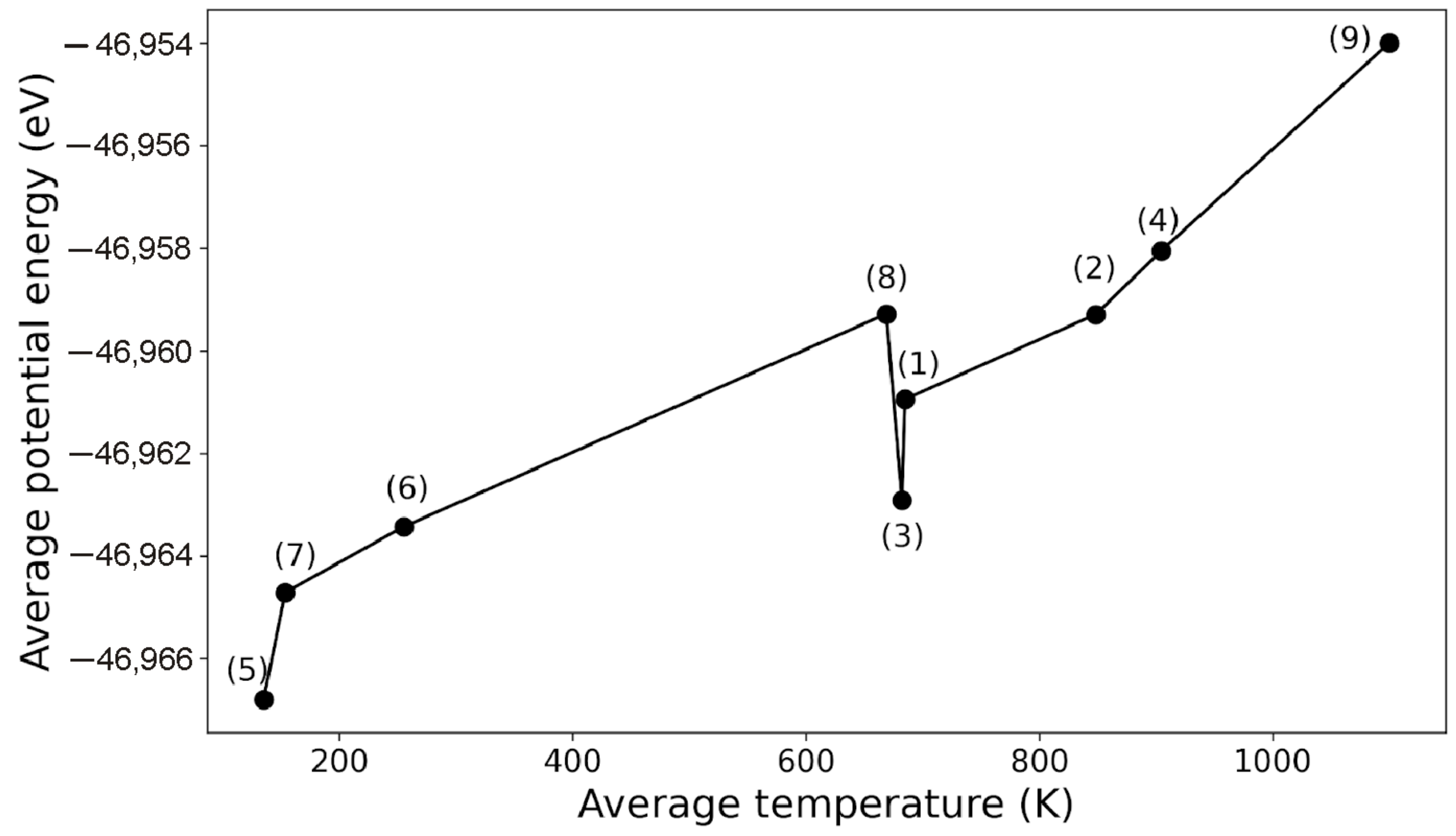
The temperature dependence of the average potential energy of perovskite LaScO
3 is not monotonic (
Figure 5). Sharp changes in the energy
U are observed at temperatures
T < 153 K and in the temperature range 669 ≤
T ≤ 685 K. In this temperature range, the highest energy
U corresponds to the state with the highest value of the electric field and the least energy appears when the field is missing. The largest value of the energy
U is also observed at
T = 1099 K at the maximum value of the electric field strength. The behavior of a proton in a system essentially depends on the temperature of the perovskite. We can say with confidence that no proton jumps are observed at temperatures below 400 K; the proton vibrates around the oxygen atom near which it was originally located. In the temperature range of 600–800 K, random proton jumps are observed between oxygen atoms. Most of the proton jumps are observed at the temperatures above 800 K, when the proton has a sufficient speed to overcome the potential barrier. A visual representation of the proton motion in the LaScO
3 perovskite is provided in
Supplementary data. This film representation shows the behavior of the proton in the calculation 1, when the temperature in the model was 685 K.
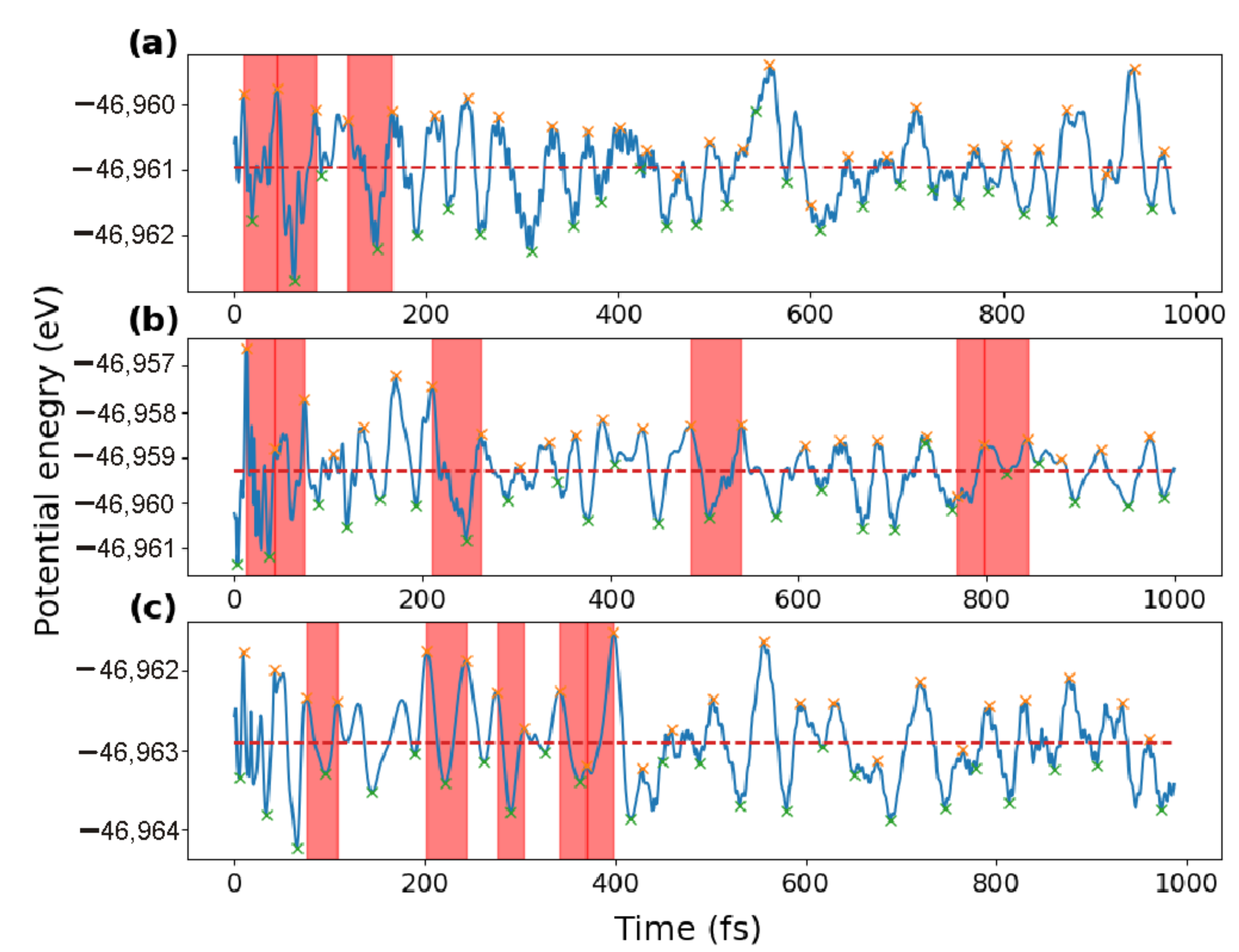
The behavior of a proton in a perovskite system in the temperature range of 680–850 K makes it possible to distinguish the time interval, during which the proton jumps to a neighboring oxygen atom, or to another place of temporal localization. In the time sweep of the potential energy, such an interval is always included between the local extrema of the function
U(
t).
Figure 4 shows the
U(
t) functions obtained in the series 1–3, defined by
Table 1. The time intervals of the proton jump to a new sedentary position are highlighted in
Figure 6 with red stripes. In all cases, the duration of the interval for the jump did not exceed 100 fs, i.e., 100 time steps. Obviously, during such a time interval, the proton completely “forgets” the initial direction of its velocity. The further speed of the proton is determined by the interatomic interaction, temperature, and, in part, the direction of the electric field strength. The electric field does not completely determine the direction of motion of the proton, but it affects the instantaneous values of the potential energy: the greater electric field results in the more significant fluctuations of the
U(
t) function. Comparison of
Figure 6a,b testifies in favor of the fact that the frequency of the proton jumps can be influenced by the direction and magnitude of its initial velocity. In particular, at a higher value of the velocity, the time intervals between the proton jumps increase. At the same time, a comparison of
Figure 4a,c shows that the number of proton jumps in the presence of an electric field is likely to decrease.
The types of possible jumps made by a proton in a perovskite are shown in
Figure 7. The directions of the proton movement are shown by light segments connecting the light circles (proton designations). In case I, the proton changes its position, passing from the oxygen atom belonging to one octahedron to the oxygen atom assigned to another octahedron, and the transition occurs between La atoms. In case II, the proton moves along only one La atom and passes from one oxygen atom to another along the edge of the octahedron, with both oxygen atoms belonging to the same octahedron. In cases III and IV, the proton also passes one La atom, but the oxygen atoms are separated by an octahedron, i.e., together they belong to three adjacent octahedra. The forward and backward paths of the proton in these cases are not equivalent due to the difference in the atomic environment. Therefore, we consider them separately, assuming that case III corresponds to a direct transition, i.e., the proton hits the plane of the octahedron, and case IV corresponds to a reverse transition.
Figure 7 shows the characteristics of proton hopping classified according to the four types defined above. The jumps of the first type can be characterized as the migration of a proton in the lanthanum plane (the plane between the octahedra). The second type of jumps takes place on the plane of scandium (in the plane of the octahedrons). The jumps of the third type include the movement of a proton from the plane of lanthanum to the plane of scandium (transition to the plane of the octahedron). The fourth type of transition expresses the exit of the proton from the scandium plane and its return to the lanthanum plane.
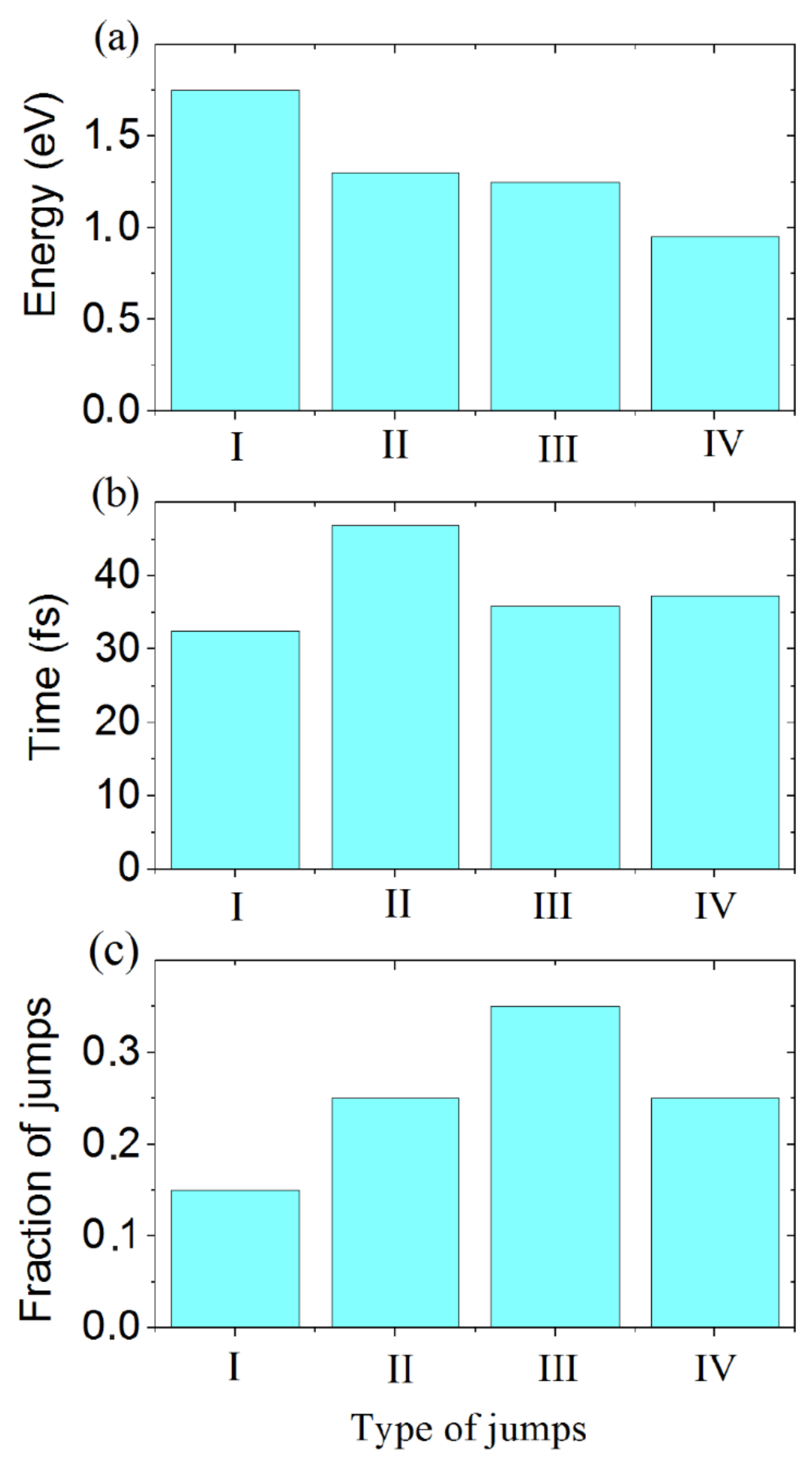
The characteristics of jumps of different types are shown in
Figure 8. Jumps of the first type are characterized by the maximum energy barrier height, the shortest jump time (since in this case the proton acquires the highest kinetic energy), and the minimum number of jumps of this type. To make a jump of the second type, the proton also needs to acquire a sufficiently large kinetic energy to overcome the second highest energy barrier. In this case, the jump time is the longest. The number of such jumps is a quarter of all jumps performed. The barrier for the jumps of the third type is slightly less than that for the jumps of the second type, and the time needed for such jumps is noticeably shorter than that for the jumps of the second type, but it is slightly longer than for the jumps of the first type. The proportion of the jumps of the third type turns out to be the largest. The jumps of the fourth type are characterized by the lowest energy barrier and the jumps time is a little longer than that for the jumps of the third type. The number of such jumps, like jumps of the second type, is a quarter of all jumps.
The migration time of a proton between the planes of lanthanum and scandium is practically independent of the direction, while the fraction of transitions into the plane of scandium is much greater than from the plane. Consequently, this jump is more beneficial for the proton than the motion in the lanthanum plane, so it prefers to move in the direction of the scandium plane. On the other hand, the movement of the proton in the scandium plane and exit from it are equally probable (the fractions of jumps of these types are the same). However, to move in the plane, the proton needs to overcome a more significant potential barrier than to exit from it. Therefore, the jump time in the scandium plane is longer.
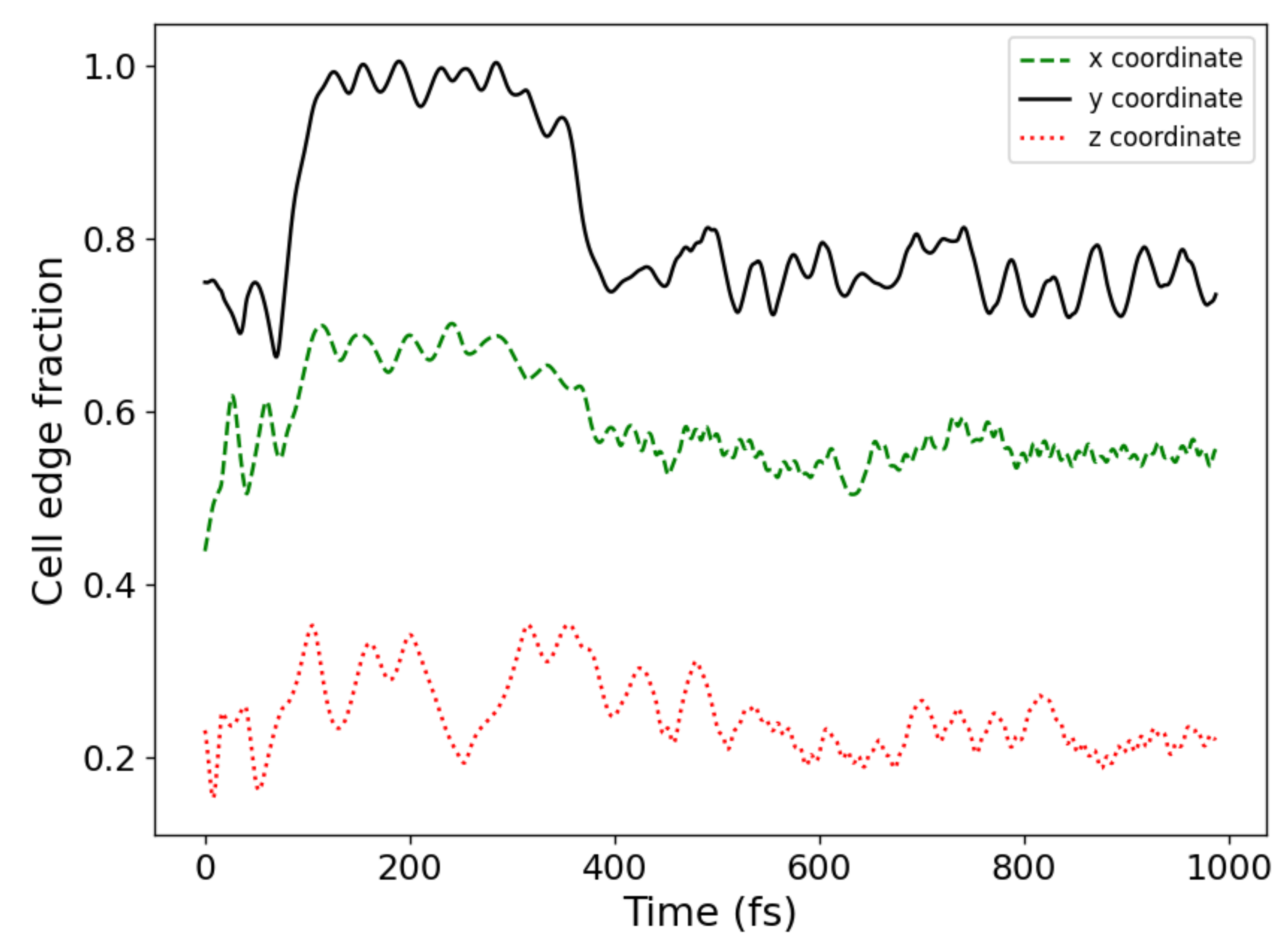
Figure 9 shows the time variation of the hydrogen run in the Cartesian coordinates from the series 3, in which 4 proton jumps were observed by the time of 400 fs. Attention should be paid to the behavior of the y coordinate, according to which it is best to diagnose jumping by a proton. A strong change in the y coordinate ends by the time of 400 fs, and further changes in this coordinate are insignificant. Small fluctuations in the
y coordinate near a constant value indicate the absence of proton jumps.
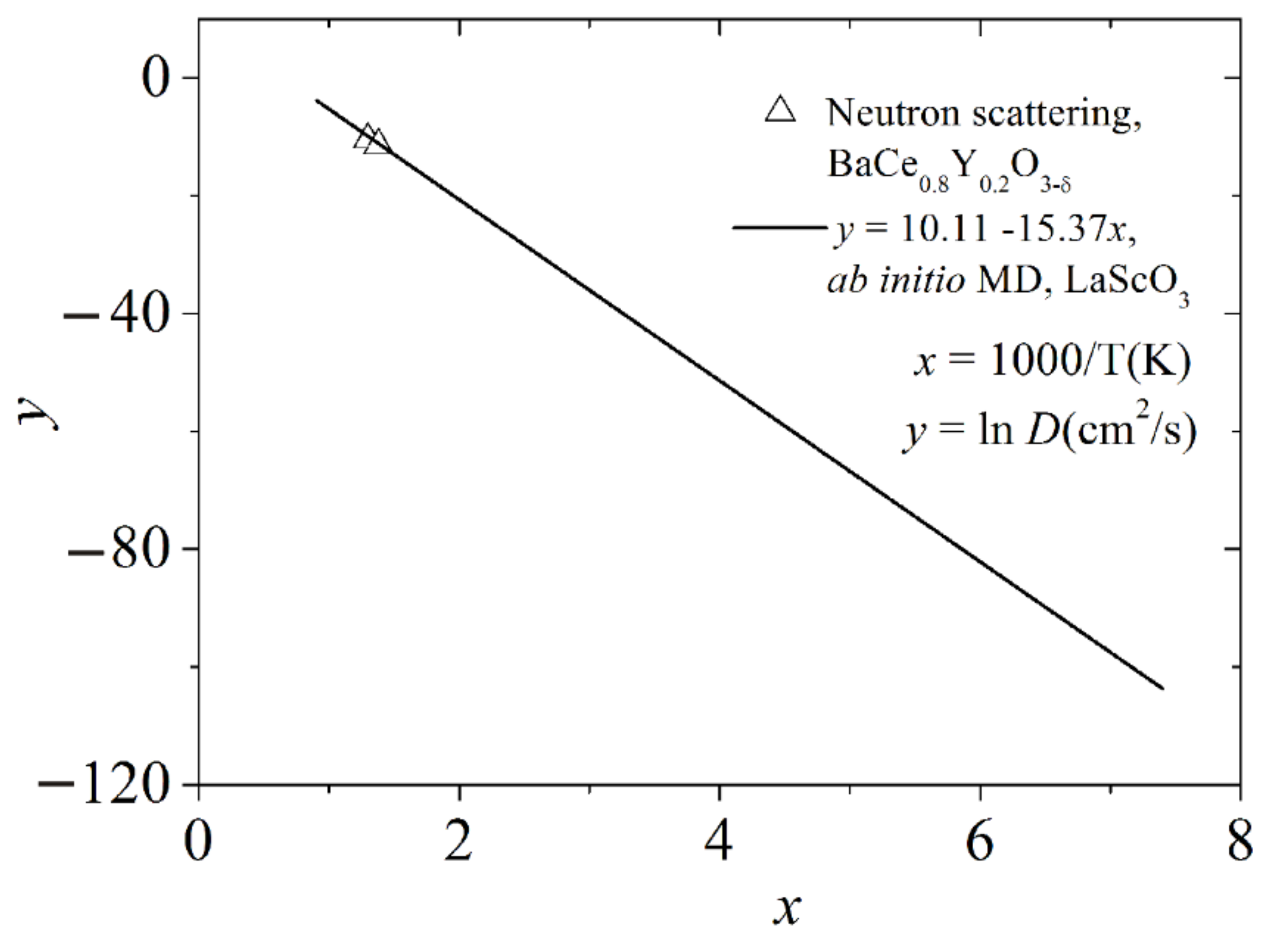
The diffusion coefficient
D was determined in terms of the mean square of the proton displacement. According to the Arrhenius law, the dependence of the diffusion coefficient on the reciprocal temperature can be represented as a linear dependence ln
D(1/
T). To calculate the average diffusion activation energy, the data presented in
Figure 8a,c were used, i.e., the average activation energy was determined, taking into account the contribution of each type of proton jump. The calculated dependence ln
D(1000/
T), showing the estimated temperature dependence of the proton diffusion coefficient in perovskite LaScO
3, is shown in
Figure 10. A similar dependence was determined for protons diffusing in yttrium-doped crystalline perovskite BaCeO
3 in a narrow temperature range (from 725 to 770 K) by the neutron scattering method [
22]. These values fit satisfactorily into the dependence ln
D(1000/
T) obtained by us for LaScO
3. Ceramics BaCe
0.8Y
0.2O
3–δ is one of the most highly conductive proton conductors. The perovskite LaScO
3 studied by us can also be attributed to this class of solid proton conductors.
Figure 11 shows the distributions of the distances between the proton and different types of atoms in the LaScO
3 perovskite system. The first peak of the “proton–lanthanum” distance distribution is split into two distinct subpeaks. This testifies the fact that, with a high probability, in addition to the nearest La atom, there are always other La atoms located at a very close distance from the proton. The highest (second) peak in this distribution shows that the majority of other La atoms are located at distances from 4 Å to 6 Å from the proton. The first peak in the “proton–scandium” distribution is not split and is well-resolved. The most probable distance from the proton to the Sc atom is 2.2 Å. The next two most pronounced peaks of this distribution are at 4.5 Å and 6 Å. It is in the vicinity of these distances that the majority of the following Sc atoms adjacent to the proton are located. The “proton–oxygen” distribution has a clearly pronounced peak at 1.0 Å, which shows that hydrogen forms a bond only with one oxygen atom, located in its field. In this case, the spectrum of distances between the proton and the rest of the oxygen atoms blurs as this distance increases. This feature of the “oxygen” spectrum is associated with a large number of O atoms in the system compared to other atoms and with their high mobility.
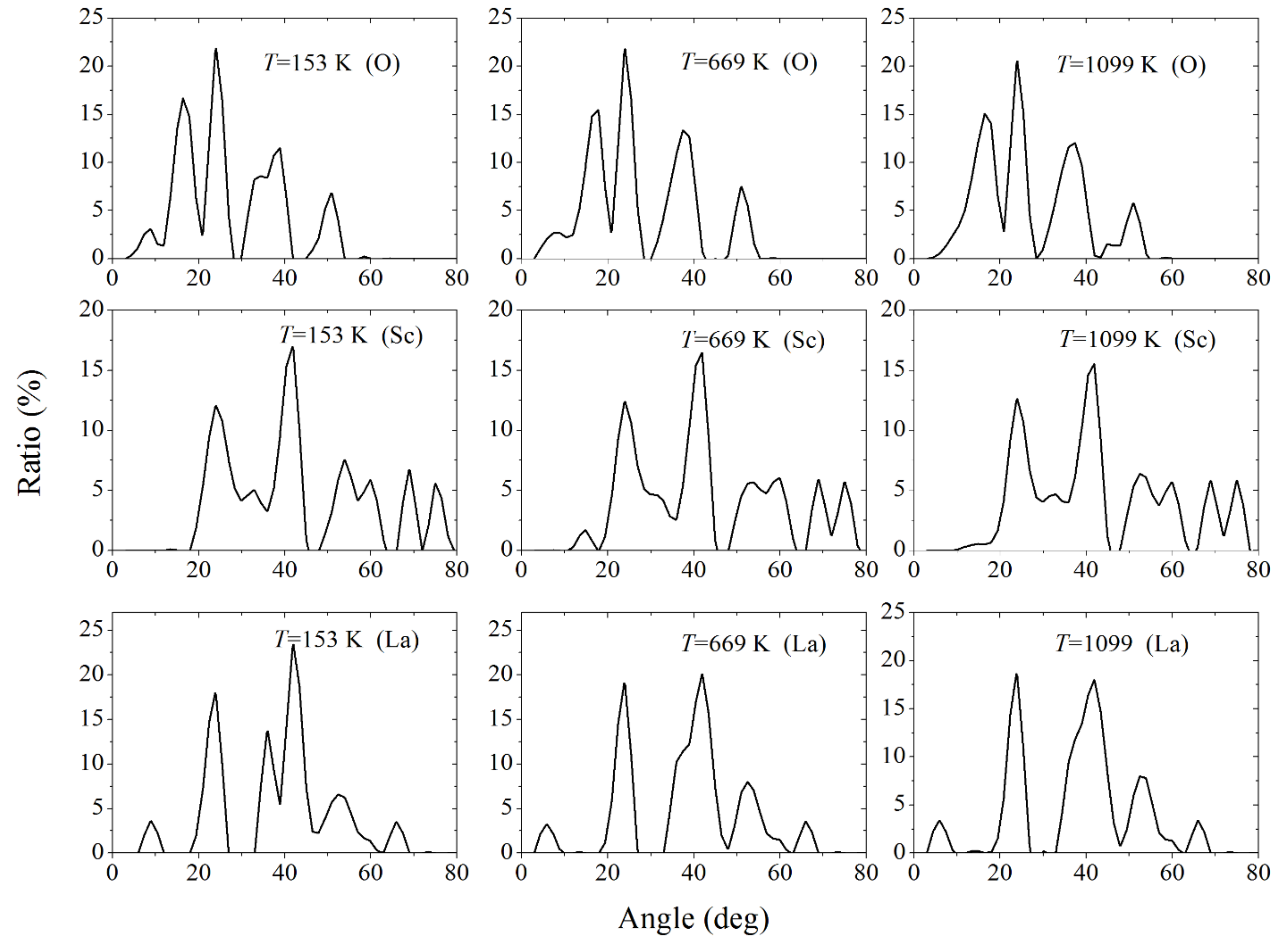
Figure 12 indirectly reflects the location of the proton by the distribution of angles that a proton forms with a pair of any atoms of the same type (i.e., O, Sc, and La). The top row of figures shows the angular distributions obtained in the absence of the electric field, and the bottom row of figures shows the same distributions when the field strength
E = 0.7 × 10
10 V/m acts in the [
10] direction in the systems under consideration. The temperature of the system in the absence and in the presence of an electric field is approximately the same (
T = 683 ± 2 K). It is seen that the corresponding angular distributions (when the angles are formed by atoms of the same type) are in good agreement with each other. The significant identity of the corresponding angular spectra indicates that the electric field has no significant effect on the proton movement in the LaScO
3 crystal.
Due to the limited space for the proton to move along the LaScO
3 crystal, which is associated with the rigid structure of this compound, an increase in the angle, at the top of which the proton is located, when the sides pass through the centers of two atoms of the same type (O, Sc, and La), means an increase in the average distance between the proton and these atoms. Due to the high mobility of the proton, it more likely interacts with the closer atoms.
Figure 12 shows that the smallest angular range is characterized by the interaction between the proton and oxygen atoms. It is this interaction that is decisive for the proton. However, the structural skeleton of a compound is not determined by oxygen, but by the metals forming this compound, i.e., Sc and La. The angular range of interaction of the proton with La is less than that with Sc. Therefore, it is easier to regulate the physical properties of the LaScO
3 compound by replacing La (rather than Sr) atoms with other atoms.
As can be seen from
Figure 13, the situation with the distribution of preferential interactions does not change, as the temperature of the system under consideration increases from 153 K to 1099 K. Even at the highest temperature, as before, the smallest angular range is observed for the proton interactions with oxygen atoms, and the largest one is for that with Sc. In all cases considered, the shape of the angular spectra does not undergo too large changes as the temperature of the system increases. Nevertheless, for the oxygen and lanthanum subsystems, the disappearance or the appearance of separate subpeaks in the angular distribution is observed at the temperature variation. Such changes are less pronounced for the scandium subsystem. Since the shape of the angular distribution characterizes indirectly the interaction between the proton and the atoms selected in the compound, the resulting response of the system to an increase in temperature means that the replacement of La by other atoms should create a corresponding effect at high temperatures.
4. Discussion
The proton conduction is an Arrhenius-type thermal activated process, phenomenologically reminiscent of a polaron-type electronic conductivity. The proton jump times can be accurately represented by the proton polaron model [
16]. Conceptually, a proton satisfies the definition of a polaron, since it can be represented as a trapped charge in an elastic crystal field. The polaron nature of proton conductivity was potentially proposed in [
23]. However, such a concept as a “proton polaron” has not yet become widespread in the study of the conductivity in perovskites, including the structures of the ABO
3 type.
The mechanism for the appearance of a polaron in an ionic crystal with an implanted proton is obvious. Longitudinal optical oscillations in such a system occur when positive and negative ions move in opposite directions. As a result of such oscillations, regions of excess (relative to the equilibrium value) charge of different signs alternating in space arise in the crystal. In the area where the proton is located, an excess of negative charge will most likely appear. It is clear that in such a system, there must be an interaction between the proton and longitudinal optical phonons. Here, the notion of a “polarization cloud” that moves along with the proton becomes appropriate. This is how a new quasi-particle, the polaron, appears. Consequently, the problem of the polaron, and not the “bare” proton, comes to the fore. Moreover, the residual interaction between the polaron and phonons is weak. A polaron drags a region of lattice distortion behind it, so its effective mass is greater than the mass of a “bare” proton, and the energy, on the contrary, is less than that of a “bare” proton. If a proton creates a deformation, which linear dimensions exceed the lattice constant, one can speak of a large-radius polaron. Otherwise, we are dealing with a small-radius polaron.
In this paper, we consider the proton–phonon interaction in the structure of the ABO3 perovskite in the form of a polaron. In addition to the aforementioned facts, we analyze the problem arising with the control of the proton conductivity in the LaScO3 perovskite using an external electric field. The path of the proton migration in the perovskite structure is determined not by the direction of the field strength, but by the elastic deformations of the crystal lattice of this compound. The mass of the La atom is 3.1 times the mass of the Sc atom. Therefore, the La-O valence phonon mode has a lower energy compared to the Sc-O mode. Consequently, in the LaScO3 perovskite, the thermally activated La-O mode acts as the main driving force for the interaction of the low-energy phonon mode and the diffusion mode of protons. Polarons appear due to the deformation of the crystal lattice. Polaron states are characterized by distinct absorption peaks in the visible (VIS) and near-IR (NIR) ranges of the optical absorption spectrum. Dielectric spectroscopy is an important tool in the study of polaron states. In particular, the electrical transition between polaron states largely determines the temperature-dependence of the relaxation frequency for the DC electrical conductivity.
When the crystal lattice contracts, the protons slow down. As the temperature rises, protons resist pressure more and more and retain a higher diffusion coefficient. With the increasing pressure on the sample, the activation barrier for proton diffusion increases. The activation energy for proton diffusion depends on the degree of the lattice deformation, which in turn is determined by the elastic properties [
24]. Proton transfer is facilitated by cooperative processes such as lattice vibrations. In perovskites of the ABO
3 type, the level of proton conductivity depends on the nature of the atoms in this compound. Typically, the proton conductivity increases as the elements A and B become less negative. The proton conductivity in perovskite can be increased by increasing the number of oxygen vacancies, i.e., by doping. In particular, the formation of oxygen vacancies is facilitated by the substitution of Sr for La in the LaScO
3 perovskite. However, this way of increasing the proton conductivity is associated with fundamental limitations. First, the formation of an oxygen vacancy is associated with the breaking of the oxygen–metal (O–M) bond, which requires a certain amount of energy. Second, the formation of an electrically neutral vacancy is associated with the reduction of neighboring cations [
25], and, therefore, with changes in the chemical energy. Third, at an excessive number of oxygen vacancies, the stability of the compound containing them can be violated [
26]. Thus, only a limited number of oxygen vacancies can be created in a perovskite of the ABO
3 type.
The perovskites based on rare earth elements demonstrate nontrivial electrical properties, such as metal-insulator transition, high-temperature superconductivity, etc. [
27]. The existence of the tunable bandwidth of the materials is explained by the variations in localized and delocalized electron patterns.
The technology of high-performance solar cells production requires uniform large-area perovskite films obtained by the crystallization process [
28]. It should be noted that the main problems of high-efficiency solar cells are the high production cost and decrease in their efficiency with an increase in the size of the solar module [
29,
30].
Polymer conductors have their own criteria for proton conductivity. Acidic hydrophilic groups often provide high proton conductivity of a compound (proton polymer conductor) with a supramolecular network [
31]. For the effective work of the proton polymer conductors, it is necessary to achieve their significant acidity, i.e., high degree of protonation of hydrophilic groups. The acidity index is the main factor in the synthesis of the polymer conductor with good proton-conducting ability.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}