

VAPPD: Visual Analysis of Protein Pocket Dynamics

 , ,
, , 
Abstract
:1. Introduction
- A coding representation based on the shape combined with topological features of protein molecular pockets is proposed to improve, to some extent, the accuracy of high-dimensional pocket similarity calculations.
- A novel high-dimensional pocket similarity calculation method based on P2V-DTW is proposed to solve the correlation calculation of unequal length sequences in high-dimensional data.
- A progressive visual analysis method of protein molecular pockets is adopted, with specific consideration of its multi-scale properties (in time and space). This method can explore the stability, similarity, and physicochemical properties of high-dimensional pockets, and discover potential allosteric pockets.
2. Related Work
2.1. Pocket Calculation
2.2. Allosteric Site Prediction
2.3. Pocket Visualization
3. Requirements
- R1: Pocket stability is used as a key feature to determine the priority of structure-based drug design. Track the changes of pockets in molecular dynamics simulation, including appearance, frequency, volume change, and disappearance, and select molecular pockets with higher stability.
- R2: Pockets with strong correlation with orthosteric pockets may be potential allosteric pockets, which are used to design allosteric compounds. Solving the problem of different time sequences and different alpha sphere number sequences will help to retain more pocket features, calculate the correlation between high-dimensional pockets, and perform better in the prediction of alternative pockets.
- R3: The physical and chemical properties of dynamic molecular pockets can be presented, which supports the cross-validation of pocket shape and pocket features. At the same time, biologists hope to obtain drugs that act on allosteric sites, and the spatiotemporal exploration of the physical and chemical properties of pockets is conducive to the screening of allosteric drugs.
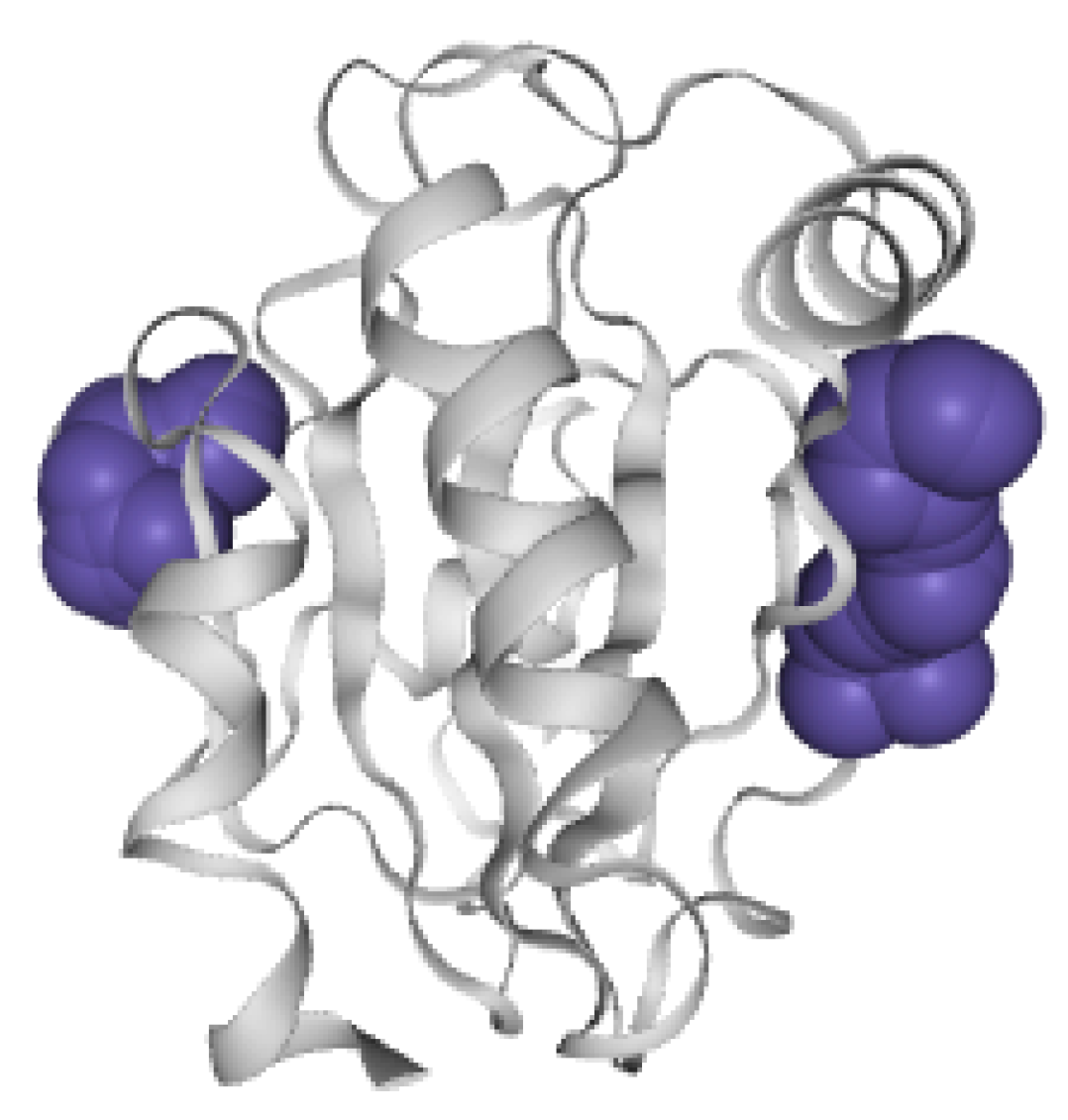
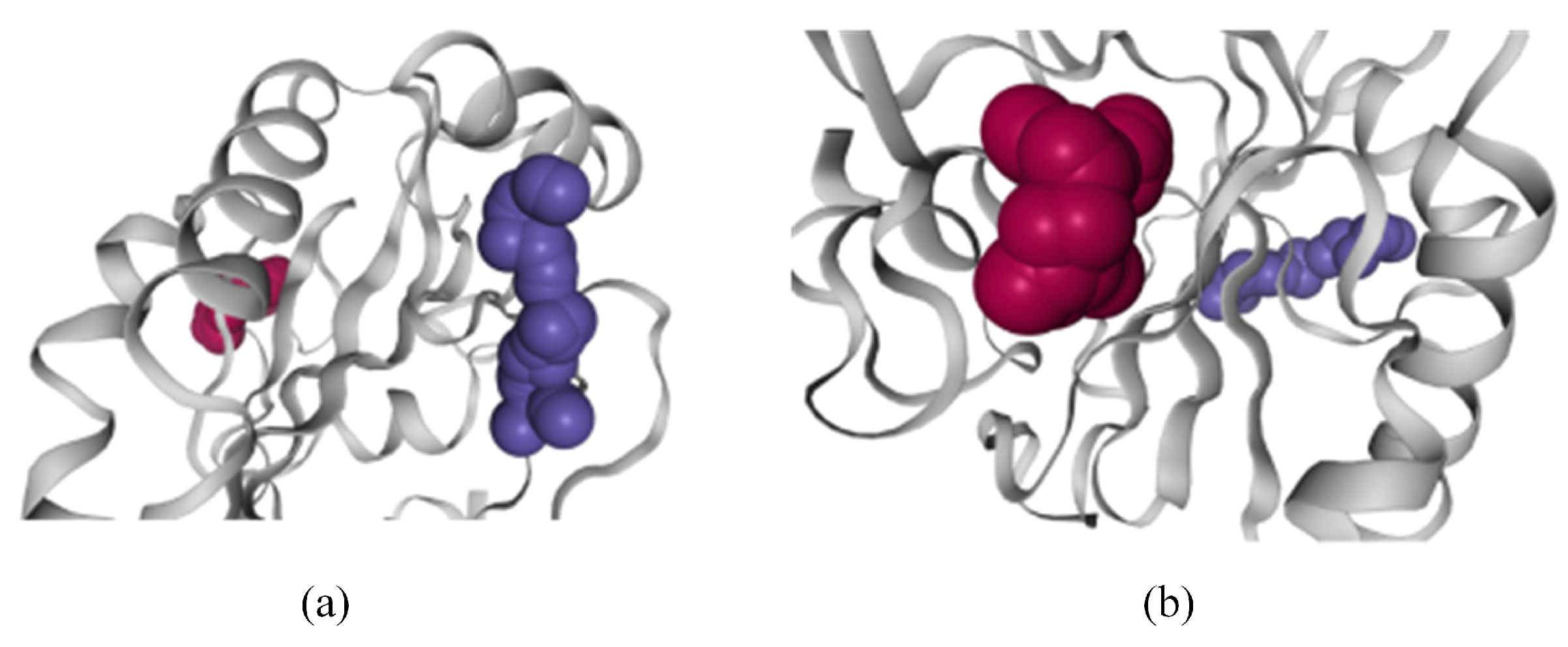
- R4: Observe the spatial shape and position of molecular pockets. Biologists tend to perceive the real shape of pockets. The 3D display is an effective method by which to discover allosteric pockets and explore the spatial features of allosteric pockets, helping biologists establish the visual perception of molecular pockets.
4. VAPPD Overview
5. Dynamic Pocket Feature Extraction
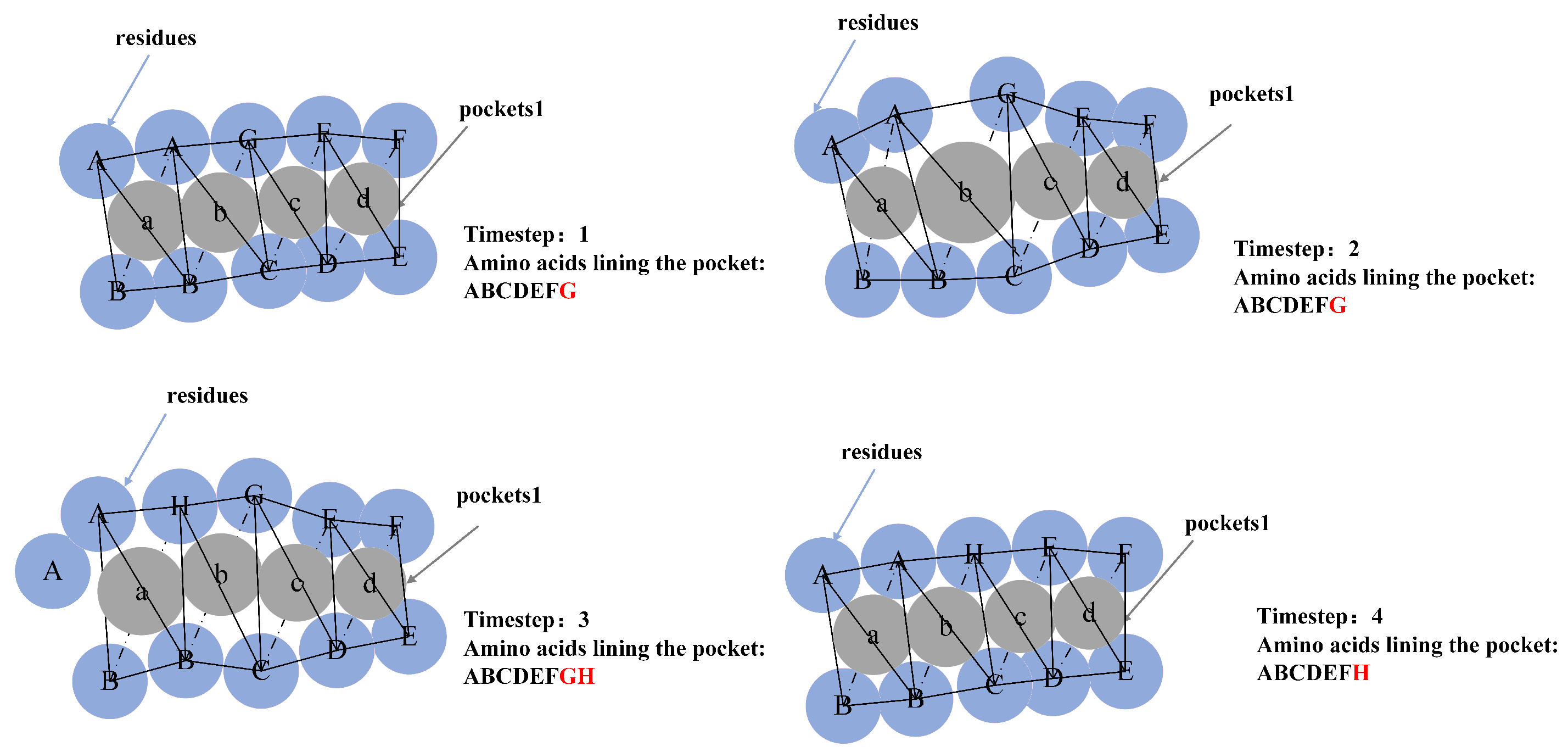
5.1. Molecular Pocket Extraction
5.2. Pocket Stability Calculation Based on Alpha Spheres
5.2.1. Pocket Hydrophobicity and Polarity Calculation
5.2.2. Pocket Volume Sequence Extraction
5.2.3. Pocket Stability Representation
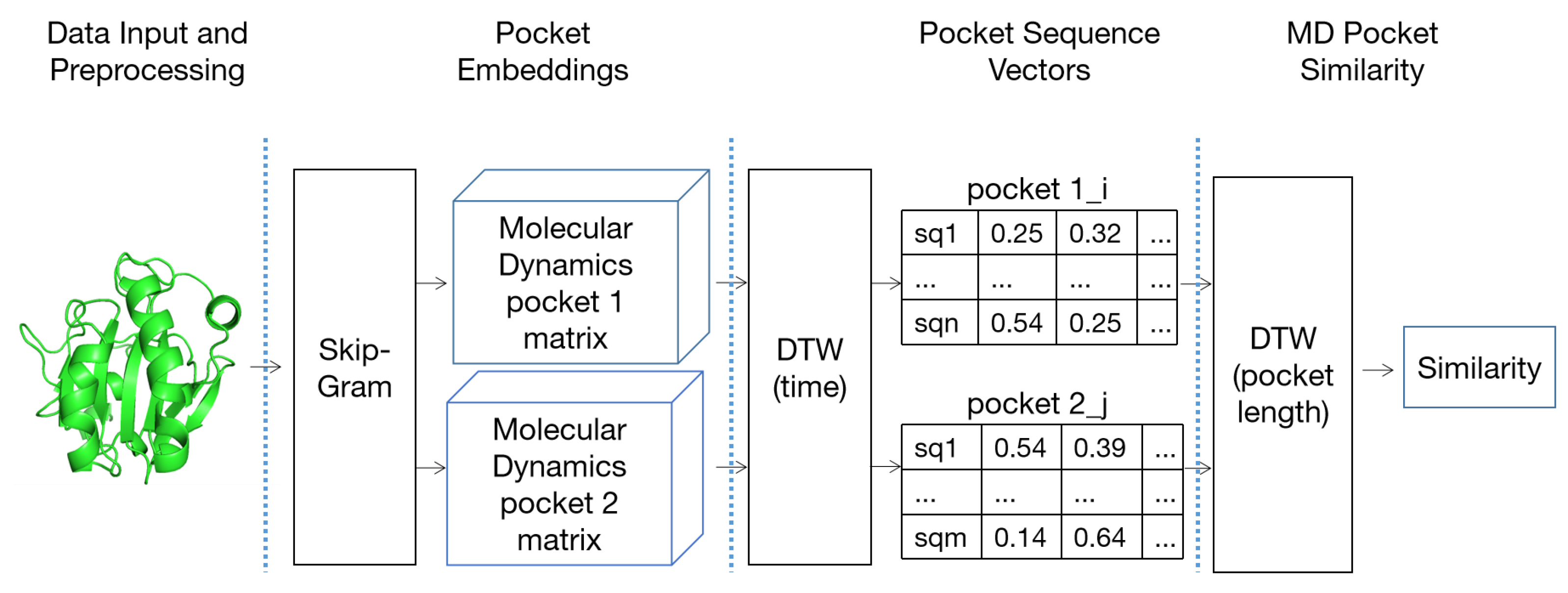
5.3. High-Dimensional Pocket Similarity Calculation Based on P2V-DTW
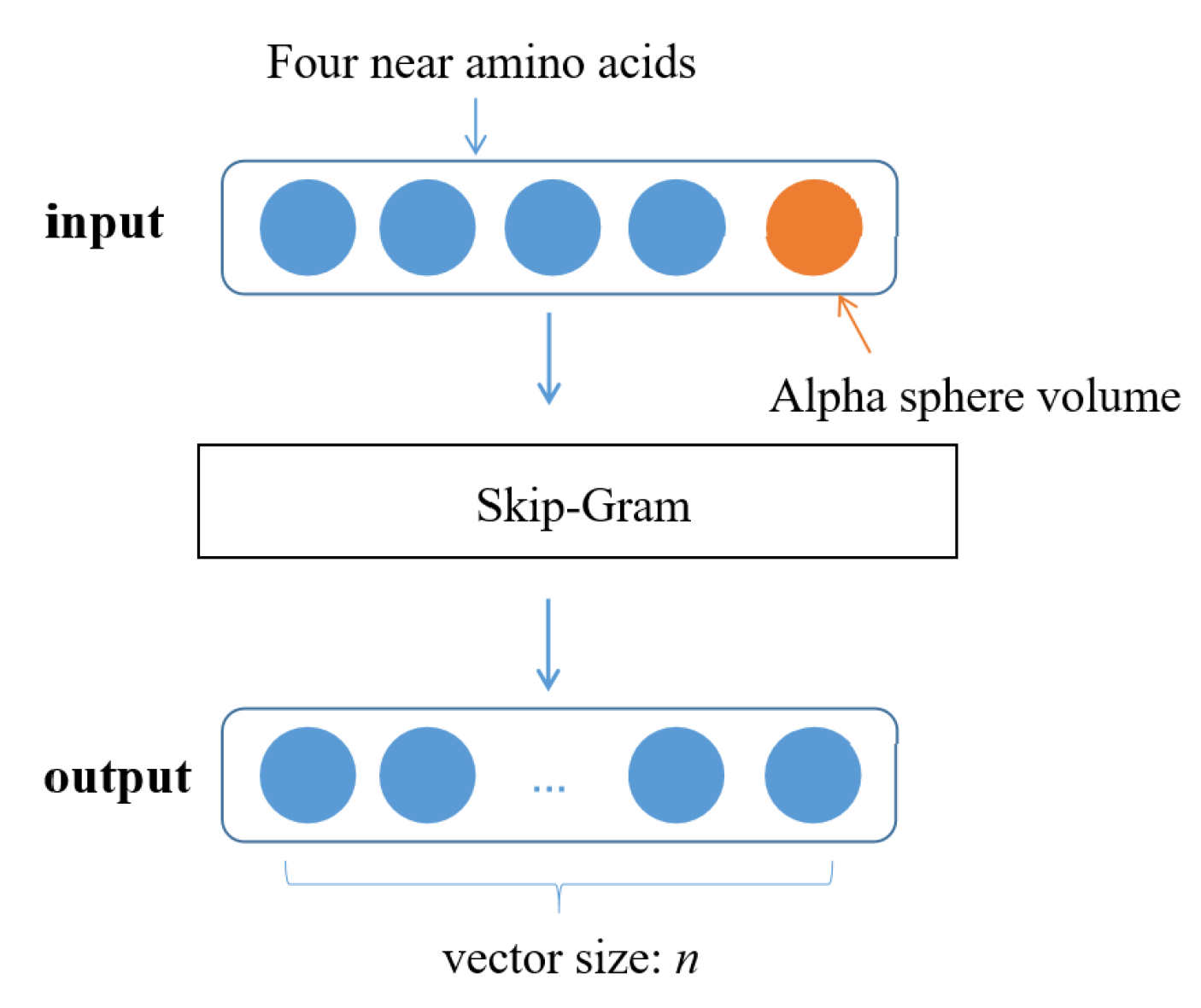
5.3.1. Pocket Word Vectorization Based on Word2Vec
5.3.2. High-Dimensional Pocket Correlation Calculation Based on P2V-DTW
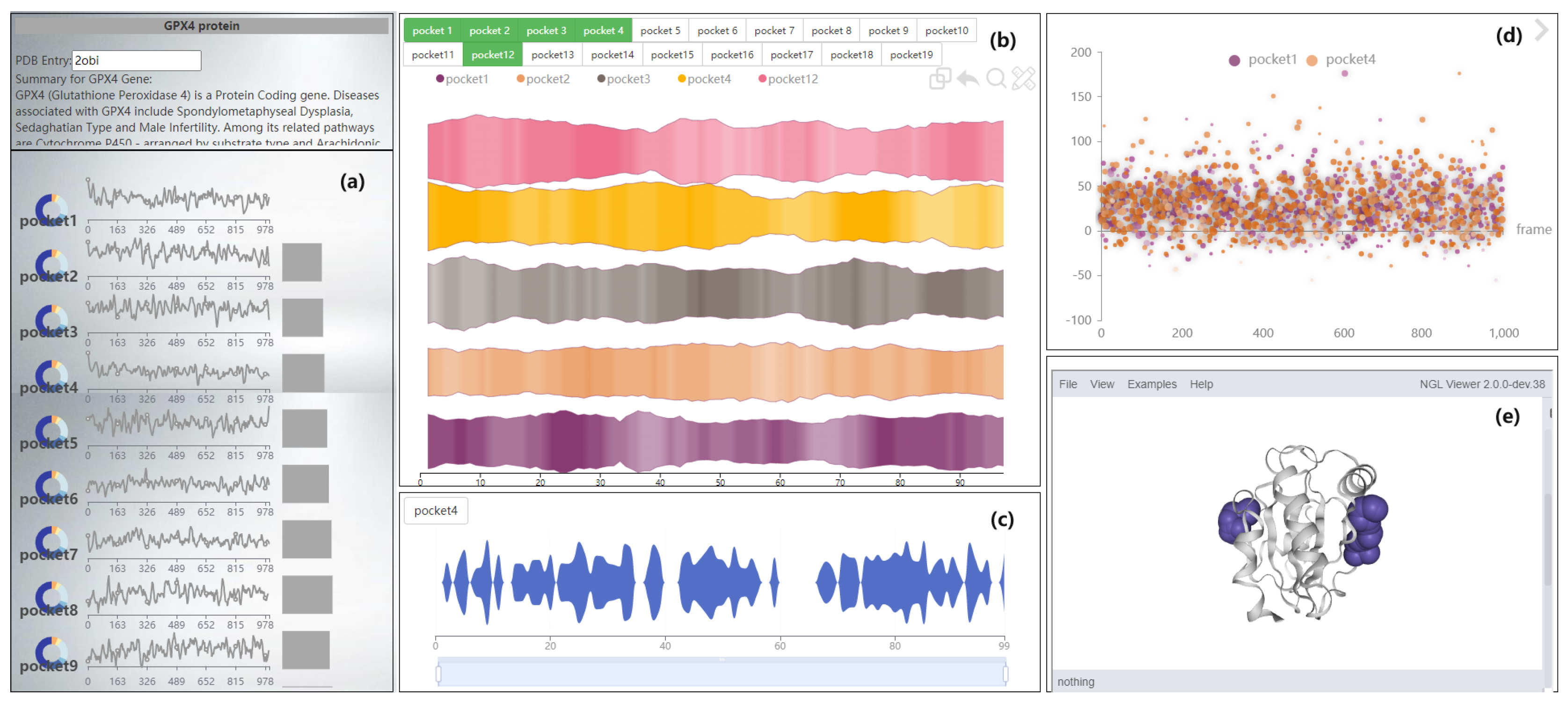
6. Progressive Visual Analysis of Protein Molecular Pockets
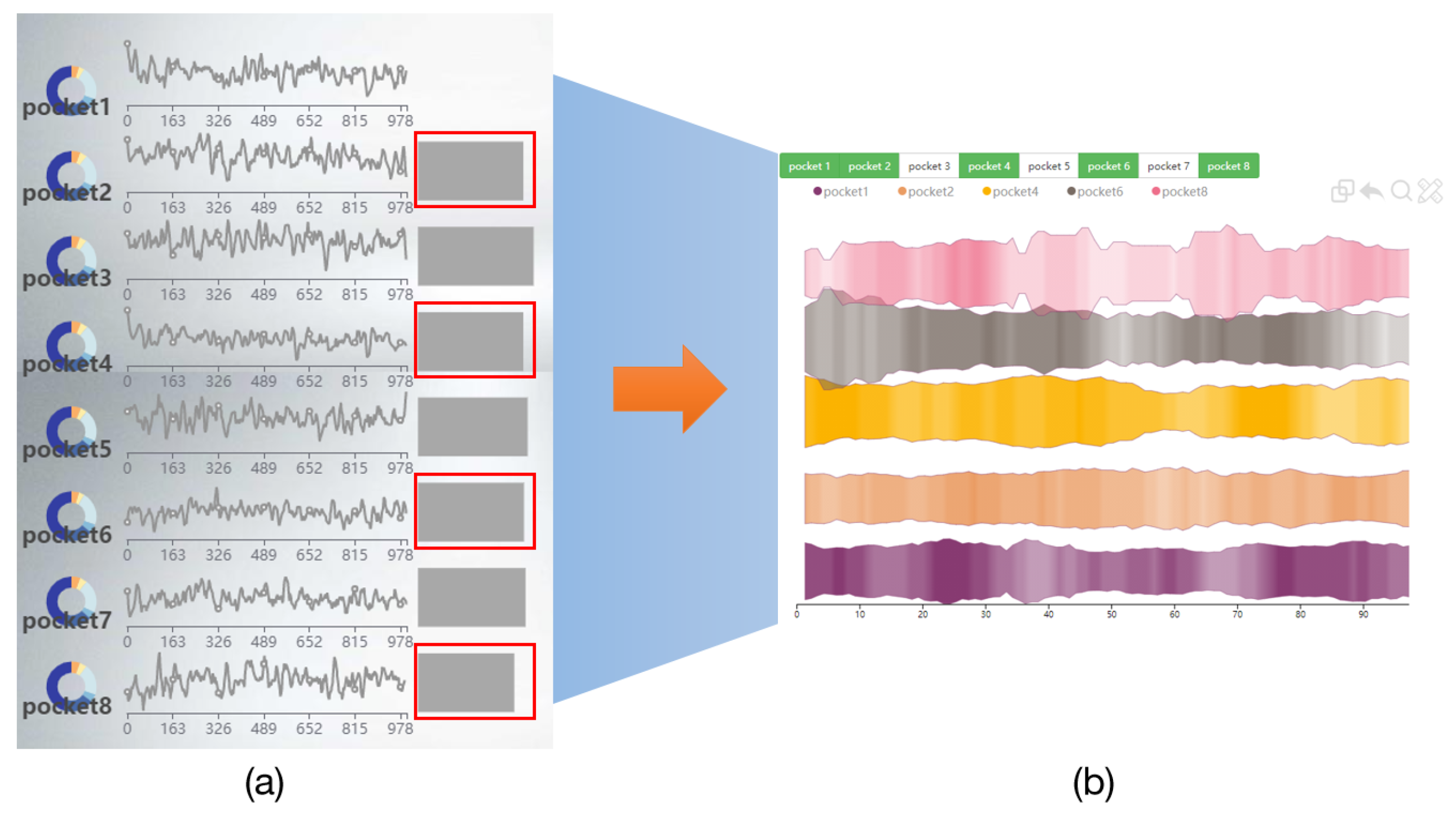
6.1. Navigation View of GTS-TP
6.2. Pocket Comparison View
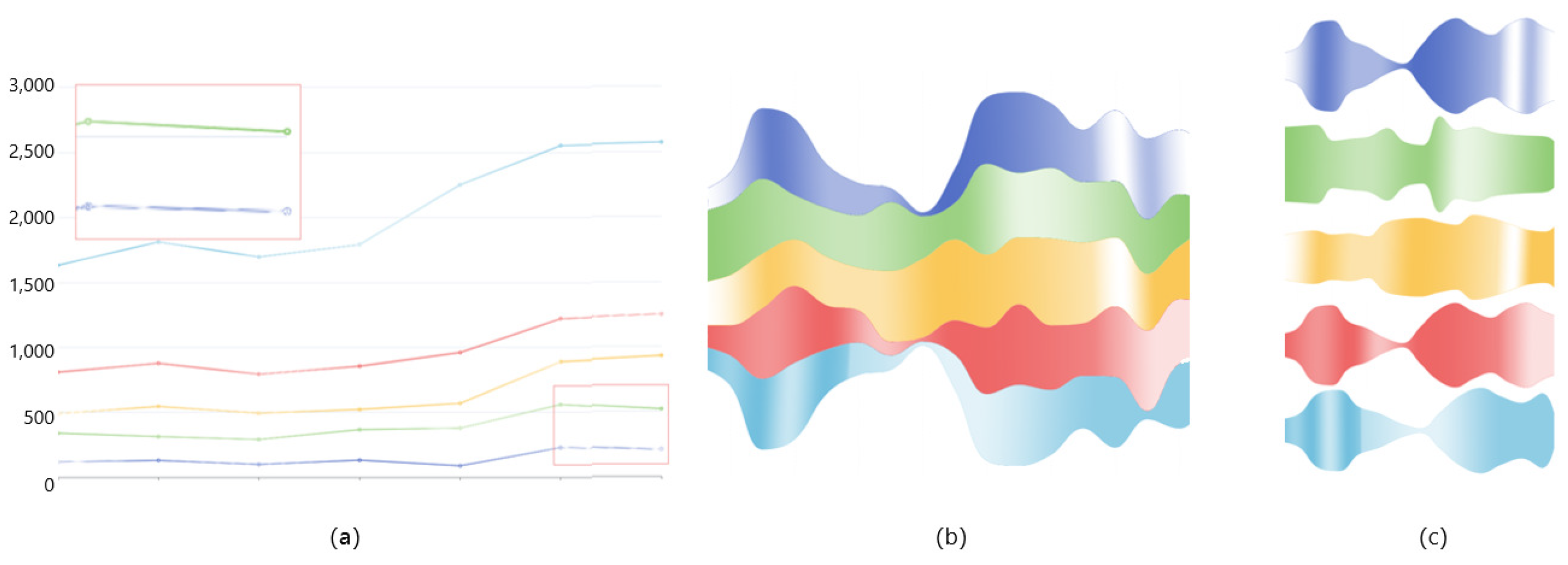
6.2.1. Comparison of Three Visualization Methods
6.2.2. Deformed River Map of GTS-CP
6.2.3. Magnifying Glass View of LTS-SP
- The P2V-DTW algorithm is provided in the pocket comparison area to compare pockets, and users are also supported to manually switch pockets for comparison.
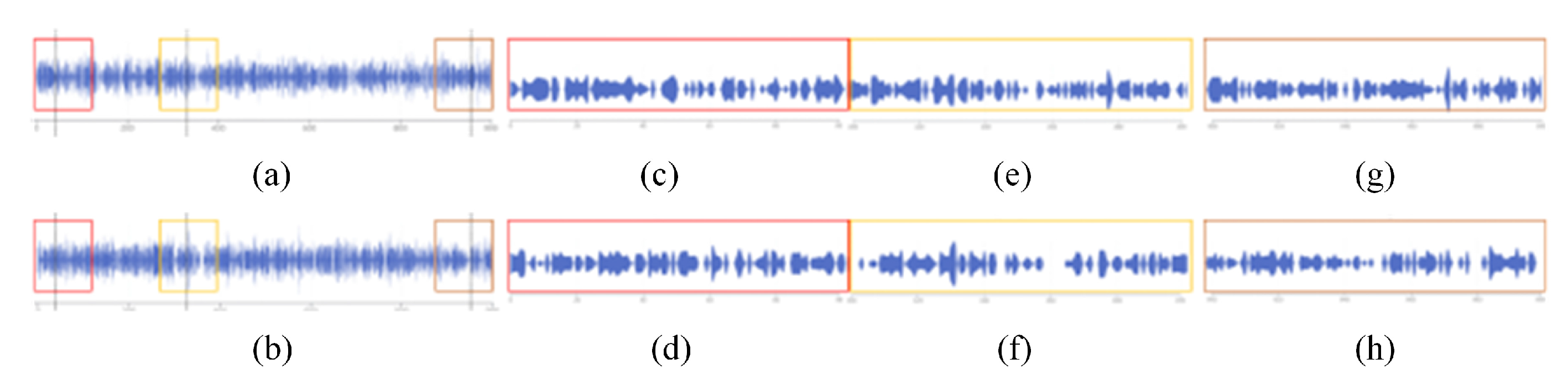
- Provide a method for detecting subtle changes in a molecular dynamics pocket after compression. When comparing pockets, the user wants to get an overview of two pockets, and compare more details. A method of slicing the data is provided to help users to better compare the pockets, and a focus river chart is used to present the sliced data. When the magnifying glass component is activated, the focus river chart follows the mouse to zoom in and out. Clicking the left mouse button will pause or start updating the data (Figure 10).
- Support for adjusting the extent of data display when observing subtle features of pockets. Interaction has been added to the focal river chart to adjust the size of the data slices by sliding the mouse wheel up to increase the magnification and down to decrease the magnification.
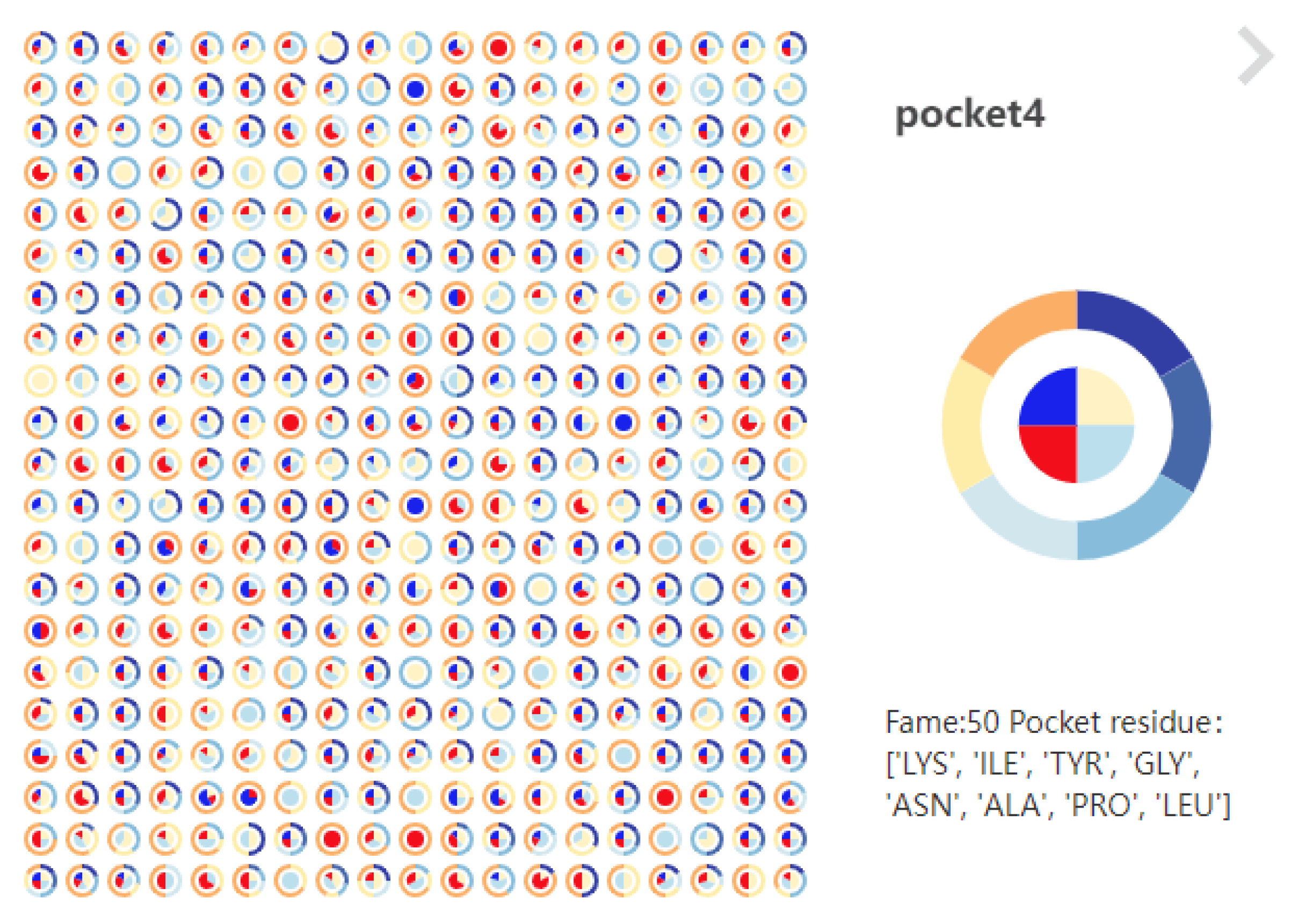
- Support for cross-analysis of pocket morphological characteristics and pocket physicochemical properties. Click on any specific moment in the focus river chart to see the pocket physicochemical properties and the proximity of amino acids at the moment.
6.3. Other Feature Views
6.3.1. Pocket Scatter Plot of GTS-CP
6.3.2. Pocket Pie of LTS-SP
6.3.3. Pocket 3D View of STS-SP
7. Interactive Exploration
8. Case Study and Feedback
8.1. Case Study
8.1.1. Pocket Correlation Calculation Results of P2V-DTW
8.1.2. Progressive Visual Analysis Results of Molecular Pockets
8.2. Feedback
9. Conclusions
- (1)
- A special representation of dynamic pocket data is proposed. This method can better characterize protein molecular pockets, and the pocket code can also be used as the input data of natural language processing model.
- (2)
- An algorithm called P2V-DTW for the correlation of molecular dynamic pocket sequences is proposed. The algorithm solves the problem of correlation calculation of unequal length sequences in high-dimensional data, and can better compare the dynamic pocket characteristics of protein molecules.
- (3)
- A progressive visual analysis method of pocket feature exploration is proposed. This method has a variety of potential applications, such as the identification of normal and allosteric pockets, the identification of stable pockets, and pocket-based drug design.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozlíková, B.; Krone, M.; Falk, M.; Lindow, N.; Baaden, M.; Baum, D.; Hege, H.C. Visualization of biomolecular structures: State of the art revisited. Comput. Graph. Forum 2017, 36, 178–204. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, X.; Peng, C.; Wang, J.; Xu, Z.; Chen, K.; Zhu, W. D3Pockets: A method and Web server for systematic analysis of protein pocket dynamics. J. Chem. Inf. Model. 2019, 59, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Eyrisch, S.; Helms, V. Transient pockets on protein surfaces involved in protein-protein interaction. J. Med. Chem. 2007, 50, 3457–3464. [Google Scholar] [CrossRef]
- Laurent, B.; Chavent, M.; Cragnolini, T.; Dahl, A.C.E.; Pasquali, S.; Derreumaux, P.; Baaden, M. Epock: Rapid analysis of protein pocket dynamics. Bioinformatics 2015, 31, 1478–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Zhang, J. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Xie, J.; Wang, S.; Xu, Y.; Deng, M.; Lai, L. Uncovering the Dominant Motion Modes of Allosteric Regulation Improves Allosteric Site Prediction. J. Chem. Inf. Model. 2021, 62, 187–195. [Google Scholar] [CrossRef]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of protein allosteric and regulatory sites. Bioinformatics 2014, 30, 1314–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Meng, H.; Lai, L. Motions of allosteric and orthosteric ligand-binding sites in proteins are highly correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Gao, Z. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 1–10. [Google Scholar] [CrossRef]
- Levitt, D.G.; Banaszak, L.J. POCKET: A computer graphies method for identifying and displaying protein cavities and their surrounding amino acids. J. Mol. Graph. 1992, 10, 229–234. [Google Scholar] [CrossRef]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Weisel, M.; Proschak, E.; Schneider, G. PocketPicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007, 1, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph. 1995, 13, 323–330. [Google Scholar] [CrossRef]
- Ho, B.K.; Gruswitz, F. HOLLOW: Generating Accurate Representations of Channel and Interior Surfaces in Molecular Structures. BMC Struct. Biol. 2008, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Teresa, P.M. MSPocket: An orientation-independent algorithm for the detection of ligand binding pockets. Bioinformatics 2011, 27, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Damborsky, J. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Jie, L.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 2010, 7, 1884–1897. [Google Scholar]
- Oliveira, S.H.; Ferraz, F.A.; Honorato, R.V. KVFinder: Steered identification of protein cavities as a PyMOL plugin. BMC Bioinform. 2014, 15, 197. [Google Scholar] [CrossRef]
- Simoes, T.; Gomes, A. CavVis—A field-of-view geometric algorithm for protein cavity detection. J. Chem. Inf. Model. 2019, 59, 786–789. [Google Scholar] [CrossRef]
- Brady, G.P.; Stouten, P.F.W. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, F.; Zhang, J.; Tang, Y.; Zhao, J.; Zhou, L.; Singh, A.K. Particle-based calculation and visualization of protein cavities using SES models. IEEE J. Biomed. Health Inform. 2021, 26, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, 374–379. [Google Scholar] [CrossRef]
- Jurcik, A.; Bednar, D.; Byška, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kozlikova, B. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef] [Green Version]
- Peter, S.; Le, G.V.; Julien, M.; Tuffïry, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, 582–589. [Google Scholar]
- Manak, M. Voronoi-based detection of pockets in proteins defined by large and small probes. J. Comput. Chem. 2019, 33, 521–533. [Google Scholar] [CrossRef]
- Huang, W.; Lu, S.; Huang, Z.; Liu, X.; Mou, L.; Luo, Y.; Zhang, J. Allosite: A method for predicting allosteric sites. Bioinformatics 2013, 29, 2357–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Qi, Y.; Lai, L. Allosteric sites can be identified based on the residue–residue interaction energy difference. Proteins Struct. Funct. Bioinform. 2015, 83, 1375–1384. [Google Scholar] [CrossRef]
- Li, C.; Deng, X.; Zhang, W.; Xie, X.; Conrad, M.; Liu, Y.; Lai, L. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J. Med. Chem. 2018, 62, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wang, Q.; Liang, M.; Liu, W.; Nie, J. Molecular cavity topological representation for pattern analysis: A NLP analogy-based Word2Vec method. Int. J. Mol. Sci. 2019, 20, 6019. [Google Scholar] [CrossRef] [PubMed]
- Raina, V.; Krishnamurthy, S. Natural language processing. In Building an Effective Data Science Practice; Apress: Berkeley, CA, USA, 2022; pp. 63–73. [Google Scholar]
- Krone, M.; Kozlíková, B.; Lindow, N.; Baaden, M.; Baum, D.; Parulek, J.; Viola, I. Visual analysis of biomolecular cavities: State of the art. Comput. Graph. Forum 2016, 35, 527–551. [Google Scholar] [CrossRef]
- Lindow, N.; Baum, D.; Bondar, A.N.; Hege, H.C. Exploring cavity dynamics in biomolecular systems. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parulek, J.; Turkay, C.; Reuter, N.; Viola, I. Implicit surfaces for interactive graph based cavity analysis of molecular simulations. In Proceedings of the 2012 IEEE Symposium on Biological Data Visualization (BioVis), Seattle, WA, USA, 14–15 October 2012; IEEE: PIscataway, NJ, USA, 2012; pp. 115–122. [Google Scholar]
- Parulek, J.; Turkay, C.; Reuter, N.; Viola, I. Visual cavity analysis in molecular simulations. BMC Bioinform. 2013, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Byška, J.; Jurčík, A.; Gröller, M.E.; Viola, I.; Kozlikova, B. MoleCollar and tunnel heat map visualizations for conveying spatio-temporo-chemical properties across and along protein voids. Comput. Graph. Forum 2015, 34, 1–10. [Google Scholar] [CrossRef]
- Byška, J.; Le Muzic, M.; Gröller, M.E.; Viola, I.; Kozlikova, B. AnimoAminoMiner: Exploration of protein tunnels and their properties in molecular dynamics. IEEE Trans. Vis. Comput. Graph. 2015, 22, 747–756. [Google Scholar] [CrossRef]
- Zhao, Y.; Ge, L.; Xie, H.; Bai, G.; Zhang, Z.; Wei, Q.; Lin, Y.; Liu, Y.; Zhou, F. ASTF: Visual Abstractions of Time-Varying Patterns in Radio Signals. IEEE Trans. Vis. Comput. Graph. 2022, in press. [Google Scholar] [CrossRef]
- Krone, M.; Kauker, D.; Reina, G.; Ertl, T. Visual analysis of dynamic protein cavities and binding sites. In Proceedings of the 2014 IEEE Pacific Visualization Symposium, Yokohama, Japan, 4–7 March 2014; IEEE: PIscataway, NJ, USA, 2014; pp. 301–305. [Google Scholar]
- Masood, T.B.; Sandhya, S.; Chandra, N.; Natarajan, V. CHEXVIS: A tool for molecular channel extraction and visualization. BMC Bioinform. 2015, 16, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Han, D.; Xu, X.; Ye, K.; Nie, J. Spatiotemporal multiscale molecular cavity visualization and visual analysis. J. Vis. 2020, 23, 661–676. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. ChemInform Abstract: A View of the Hydrophobic Effect. Cheminform 2010, 33, 521–533. [Google Scholar] [CrossRef]
- Jianwen, L.; Kui, Y.; Jing, B. Savitzky CGolay smoothing and differentiation filter for even number data. J. Abbr. 2005, 85, 1429–1434. [Google Scholar]
- Sawada, S.; Itoh, T.; Misaka, T.; Obayashi, S.; Czauderna, T.; Stephens, K. Streamline pair selection for comparative flow field visualization. Vis. Comput. Ind. Biomed. Art 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Xu, S.; Min, J.; Wang, F. Ferroptosis: An Emerging Player in Immune Cells. Sci. Bull. 2021, 22, 2257–2260. [Google Scholar] [CrossRef]
- Wang, D.S.; Hayatshahi, H.S.; Jayasinghe-Arachchige, V.M.; Liu, J. Allosteric Modulation of Small Molecule Drugs on ACE2 Conformational Change upon Binding to SARS-CoV-2 Spike Protein. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 2587–2594. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description |
|---|---|
| Time_id | Pocket timestep number. |
| Pocket_id | Pocket number. |
| Hydrophilicity | Statistics of hydrophilic amino acids in molecular pockets. |
| Hydrophobicity | Statistics of hydrophobic amino acids in molecular pockets. |
| Pocket volume | Pocket volume. |
| Pocket relevance | Pocket correlation. |
| Method | Volume | GPX Topology | Vol + Topo | Volume | ACE Topology | Vol + Topo |
|---|---|---|---|---|---|---|
| P2V-DTW | - | 5 | 2 | - | 3 | 3 |
| No P2V-DTW | 4 (D3Pockets) | 4 | 4 | 8 (D3Pockets) | 4 | 4 |
| Method | Pocket 2 | Pocket 3 | Pocket 4 | Pocket 5 | Pocket 6 | Pocket 7 | Pocket 8 |
|---|---|---|---|---|---|---|---|
| P2V-DTW | 1416.6 | 1555.2 | 1416.2 | 1474.5 | 1425.6 | 1446.4 | 1294.7 |
| Similarity Rank | 3 | 7 | 2 | 6 | 4 | 5 | 1 |
| D3Pockets [2] | 0.1002 | 0.0549 | 0.0286 | 0.0323 | 0.0168 | 0.0192 | 0.0174 |
| Similarity Rank | 1 | 2 | 4 | 3 | 7 | 5 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, D.; Feng, L.; Shi, C.; Cao, L.; Li, Y.; Wang, Y.; Xu, X. VAPPD: Visual Analysis of Protein Pocket Dynamics. Appl. Sci. 2022, 12, 10465. https://doi.org/10.3390/app122010465
Guo D, Feng L, Shi C, Cao L, Li Y, Wang Y, Xu X. VAPPD: Visual Analysis of Protein Pocket Dynamics. Applied Sciences. 2022; 12(20):10465. https://doi.org/10.3390/app122010465
Chicago/Turabian StyleGuo, Dongliang, Li Feng, Chuanbao Shi, Lina Cao, Yu Li, Yanfen Wang, and Ximing Xu. 2022. "VAPPD: Visual Analysis of Protein Pocket Dynamics" Applied Sciences 12, no. 20: 10465. https://doi.org/10.3390/app122010465
APA StyleGuo, D., Feng, L., Shi, C., Cao, L., Li, Y., Wang, Y., & Xu, X. (2022). VAPPD: Visual Analysis of Protein Pocket Dynamics. Applied Sciences, 12(20), 10465. https://doi.org/10.3390/app122010465