
Enhanced Electrodesorption Performance via Cathode Potential Extension during Capacitive Deionization

Abstract
:1. Introduction
2. Experimental
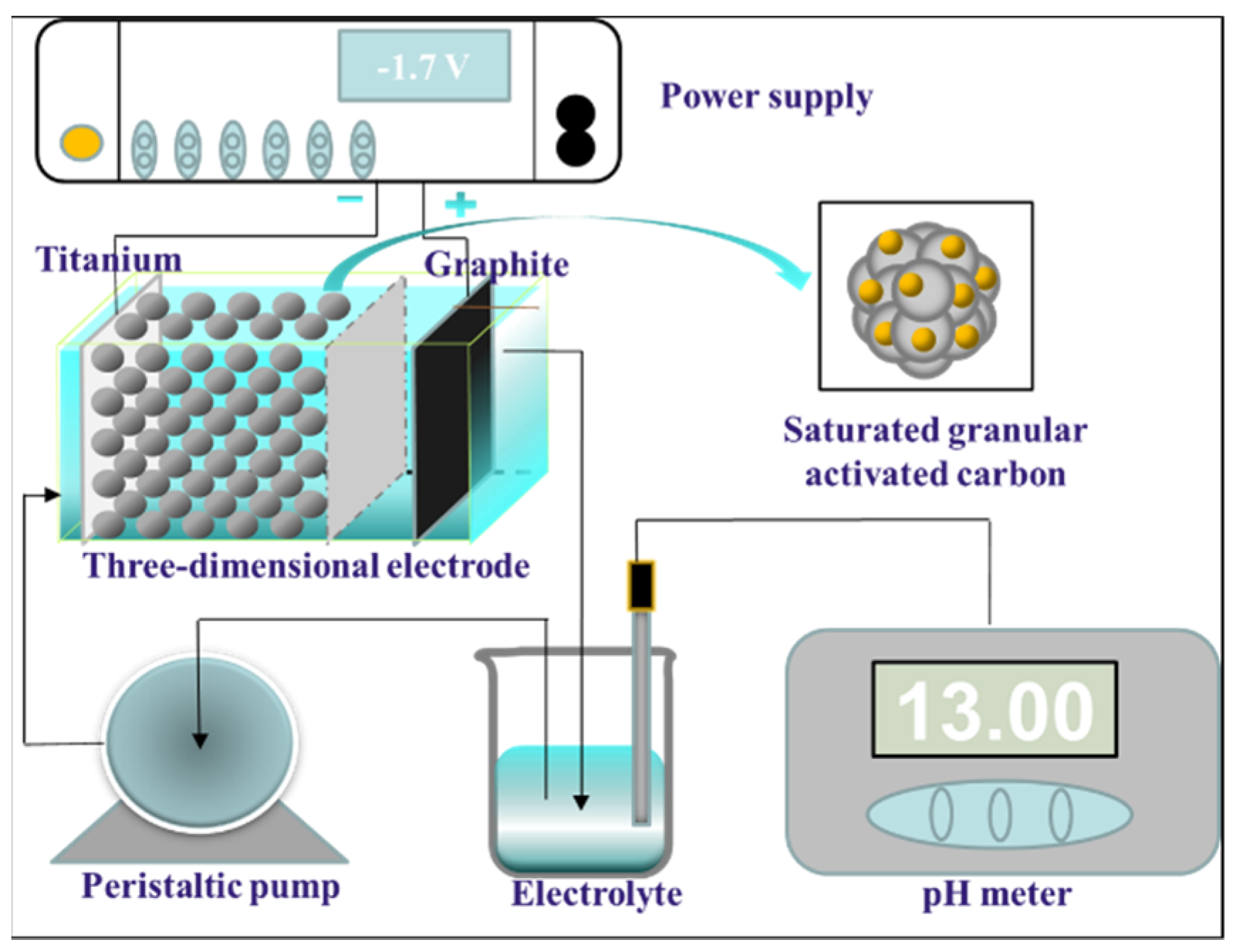
2.1. Apparatus
2.2. Materials and Methods
3. Results and Discussion
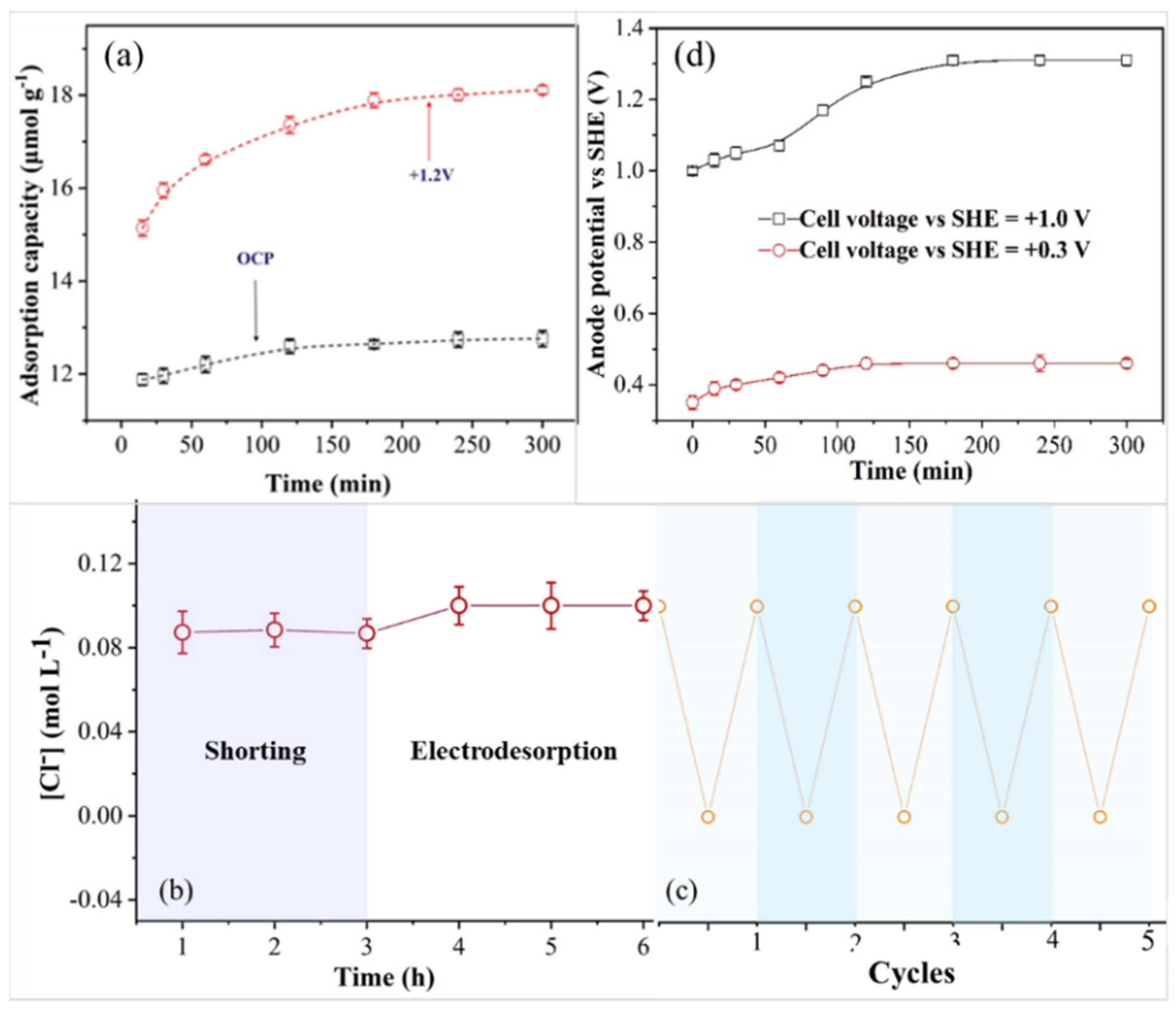
3.1. Electrosorption and Desorption Cycles
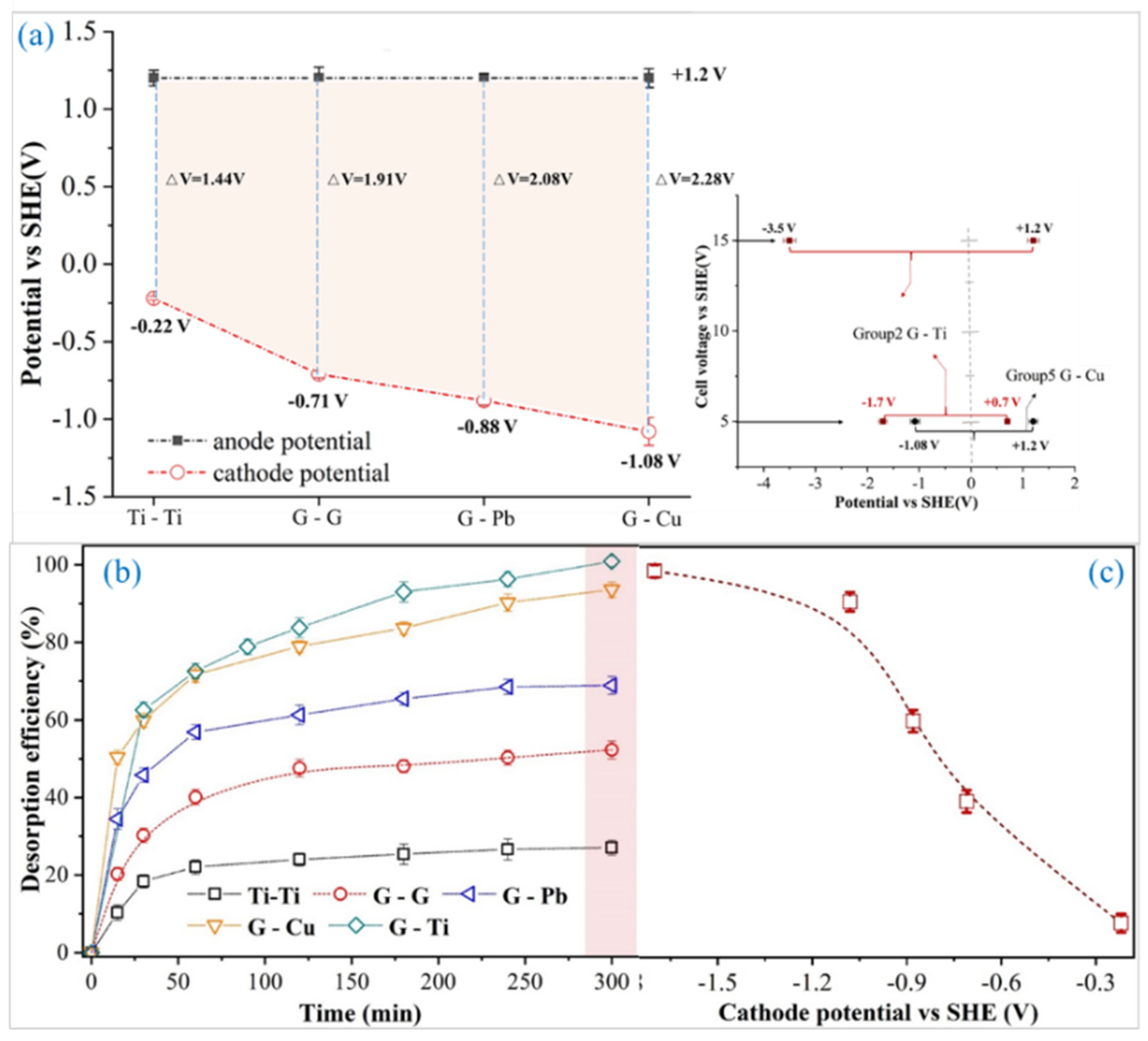
3.2. Effect of Electrode Configuration
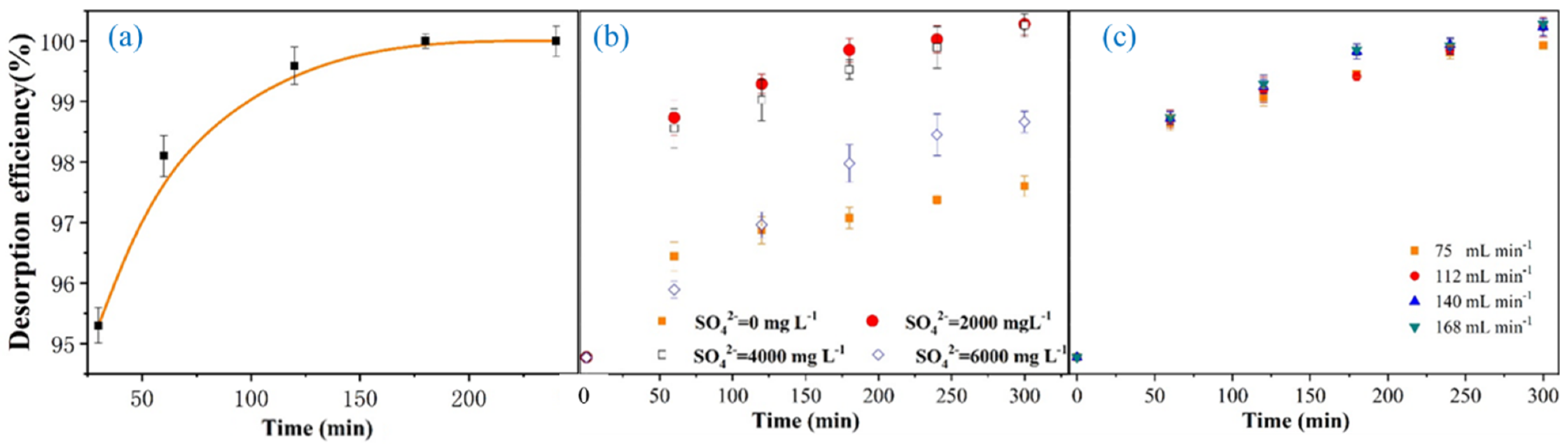
3.3. Application to Electrodesorption of SO42−
3.4. Effect of Ionic Strength
3.5. Effect of Circulating Velocity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Caudle, D.D.; Tucker, J.T.H.; Papastamataki, A. Electrochemical Demineralization of Water with Carbon Electrodes, Research Report; Oklahoma University Research Institute: Norman, OK, USA, 1966. [Google Scholar]
- Anderson, M.A.; Cudero, A.L.; Palma, J. Capacitive deionization as an electrochemical means of saving energy and delivering clean water. Comparison to present desalination practices: Will it compete? Electrochim. Acta 2010, 55, 3845–3856. [Google Scholar] [CrossRef]
- Gabelich, C.; Tran, T.; Suffet, I. Electrosorption of inorganic salts from aqueous solution using carbon aerogels. Environ. Sci. Technol. 2002, 36, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yang, Y.; Hu, Y.; Hu, M.; Wang, C. Fabrication of Graphene-Based Xerogels for removal of heavy metal ions and Capacitive Deionization. ACS Sustain. Chem. Eng. 2015, 3, 1056–1065. [Google Scholar] [CrossRef]
- Han, L.; Karthikeyan, K.G.; Anderson, M.A.; Gregory, K.B. Exploring the impact of pore size distribution on the performance of carbon electrodes for capacitive deionization. J. Colloid Interface Sci. 2014, 430, 93–99. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.; Mao, S.; Sun, Z.; Song, Y.; Zhao, R. Fabrication of porous graphene electrodes via CO2 activation for the enhancement of capacitive deionization. J. Colloid Interface Sci. 2019, 536, 252–260. [Google Scholar] [CrossRef]
- Porada, S.; Zhao, R.; Wal, A.; Presser, V.; Biesheuvel, P.M. Review on the science and technology of water desalination by capacitive deionization. Prog. Mater. Sci. 2013, 58, 1388–1442. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jo, K.; Lee, J.; Hong, S.P.; Kim, S.; Yoon, J. Rocking-Chair capacitive deionization for continuous brackish water desalination. ACS Sustain. Chem. Eng. 2018, 6, 10815–10822. [Google Scholar] [CrossRef]
- Xu, P.; Drewes, J.E.; Heil, D.; Wang, G. Treatment of brackish produced water using carbon aerogel-based capacitive deionization technology. Water Res. 2008, 42, 2605–2617. [Google Scholar] [CrossRef]
- Lado, J.J.; Zornitta, R.L.; Calvi, F.A.; Tejedor-Tejedor, M.I.; Anderson, M.A.; Ruotolo, L.A.M. Study of sugar cane bagasse fly ash as electrode material for capacitive deionization. J. Anal. Appl. Pyrol. 2016, 120, 389–398. [Google Scholar] [CrossRef]
- Suss, M.E.; Porada, S.; Sun, X.; Biesheuvel, P.M.; Yoon, J.; Presser, V. Water desalination via capacitive deionization: What is it and what can we expect from it? Energ. Environ. Sci. 2015, 8, 2296–2319. [Google Scholar] [CrossRef] [Green Version]
- Chong, L.G.; Chen, P.A.; Huang, J.Y.; Huang, H.L.; Wang, H.P. Capacitive deionization of a RO brackish water by AC/graphene composite electrodes. Chemosphere 2018, 191, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y. Capacitive deionization (CDI) for desalination and water treatment—Past, present and future (a review). Desalination 2008, 228, 10–29. [Google Scholar] [CrossRef]
- Ban, A.; Schafer, A.; Wendt, H. Fundamentals of electrosorption on activated carbon for wastewater treatment of industrial effluents. J. Appl. Electrochem. 1998, 28, 227–236. [Google Scholar] [CrossRef]
- Oh, H.-J.; Lee, J.-H.; Ahn, H.-J.; Jeong, Y.; Kim, Y.-J.; Chi, C.-S. Nanoporous activated carbon cloth for capacitive deionization of aqueous solution. Thin Solid Films 2006, 515, 220–225. [Google Scholar] [CrossRef]
- Kim, S.; Yoon, J.; Yoon, H.; Shin, D.; Lee, J. Electrochemical selective ion separation in capacitive deionization with sodium manganese oxide. J. Colloid Interface Sci. 2017, 506, 644–648. [Google Scholar] [CrossRef]
- Chao, L.; Liu, Z.; Zhang, G.; Song, X.; Lei, X.; Noyong, M.; Simon, U.; Chang, Z.; Sun, X. Enhancement of capacitive deionization capacity of hierarchical porous carbon. J. Mater. Chem. A 2015, 3, 12730–12737. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Bae, W.; Choi, J. Electrode reactions and adsorption/desorption performance related to the applied potential in a capacitive deionization process. Desalination 2010, 258, 159–163. [Google Scholar] [CrossRef]
- Lado, J.J.; Pérez-Roa, R.E.; Wouters, J.J.; Tejedor-Tejedor, M.I.; Anderson, M.A. Evaluation of operational parameters for a capacitive deionization reactor employing asymmetric electrodes. Sep. Purif. Technol. 2014, 133, 236–245. [Google Scholar] [CrossRef]
- Bayram, E.; Hoda, N.; Ayranci, E. Adsorption/electrosorption of catechol and resorcinol onto high area activated carbon cloth. J. Hazard. Mater. 2009, 168, 1459–1466. [Google Scholar] [CrossRef]
- Lado, J.J.; Pérez-Roa, R.E.; Wouters, J.J.; Tejedor-Tejedor, M.I.; Federspill, C.; Ortiz, J.M.; Anderson, M.A. Removal of nitrate by asymmetric capacitive deionization. Sep. Purif. Technol. 2017, 183, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Villar, I.; Roldan, S.; Ruiz, V.; Granda, M.; Blanco, C.; Menéndez, R.; Santamaría, R. Capacitive deionization of NaCl solutions with modified activated carbon electrodes. Energy Fuel 2010, 24, 3329–3333. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Liu, Y.; Wang, S.Y.; Peng, P.; Li, X.Y.; Xu, J.; Li, W.H. A novel integrated system of three-dimensional electrochemical reactors (3DERs) and three-dimensional biofilm electrode reactors (3DBERs) for coking wastewater treatment. Bioresour. Technol. 2019, 284, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Feng, C. Achieving high performance for electrocatalytic dechlorination with magnetic Pd/CoFe2O4 particle electrodes. Appl. Surf. Sci. 2021, 545, 149007. [Google Scholar] [CrossRef]
- Pang, T.; Wang, Y.; Yang, H.; Wang, T.; Cai, W. Dynamic model of organic pollutant degradation in three dimensional packed bed electrode reactor. Chemosphere 2018, 206, 107–114. [Google Scholar] [CrossRef]
- Chen, H.; Feng, Y.; Suo, N.; Long, Y.; Li, X.; Shi, Y.; Yu, Y. Preparation of particle electrodes from manganese slag and its degradation performance for salicylic acid in the three-dimensional electrode reactor (TDE). Chemosphere 2019, 216, 281–288. [Google Scholar] [CrossRef]
- Cho, S.; Kim, C.; Hwang, I. Electrochemical degradation of ibuprofen using an activated-carbon-based continuous-flow three-dimensional electrode reactor (3DER). Chemosphere 2020, 259, 127382. [Google Scholar] [CrossRef]
- Xu, L.N.; Zhao, H.Z.; Ni, J.R. Effect of cathode material on electrolytic treatment of Acid Orange 7 by a three-phase three-dimensional electrode reactor. Huan Jing Ke Xue 2008, 29, 942–947. [Google Scholar]
- Lv, G.; Chen, Y.; Yang, T.; Li, J. Electrocatalytic Oxidation Removal of Phenol from Aqueous Solution with Metal Oxides Doped Carbon Aerogel. J. Braz. Chem. Soc. 2017, 29, 689–694. [Google Scholar] [CrossRef]
- Oda, H.; Nakagawa, Y. Removal of ionic substances from dilute solution using activated carbon electrodes. Carbon 2003, 41, 1037–1047. [Google Scholar] [CrossRef]
- Gaikwad, M.S.; Balomajumder, C. Tea waste biomass activated carbon electrode for simultaneous removal of Cr(VI) and fluoride by capacitive deionization. Chemosphere 2017, 184, 1141–1149. [Google Scholar] [CrossRef]
- Ye, W.; Zhang, W.; Hu, X.; Yang, S.; Liang, W. Efficient electrochemical-catalytic reduction of nitrate using Co/AC0.9-AB0.1 particle electrode. Sci. Total Environ. 2020, 732, 139245. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lu, Y.; Li, X.; Lu, Y.; Zhu, G.; Hassan, M. Tertiary denitrification and organic matter variations of secondary effluent from wastewater treatment plant by the 3D-BER system. Environ. Res. 2020, 189, 109937. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, F. Analysis of electrodes matching for asymmetric electrochemical capacitor. J. Power Sources 2009, 194, 1184–1193. [Google Scholar] [CrossRef]
- Laxman, K.; Gharibi, L.A.; Dutta, J. Capacitive deionization with asymmetric electrodes: Electrode capacitance vs electrode surface area. Electrochim. Acta 2015, 176, 420–425. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, L.; Shu, Y.; Li, Q.; Liu, M. Chemical bond between chloride ions and surface carboxyl groups on activated carbon. Colloid Surf. A Physicochem. Eng. Asp. 2017, 530, 53–59. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, L.; Liu, M.; Shu, Y.; Li, Q. Capacitive deionization of chloride ions by activated carbon using a three-dimensional electrode reactor. Sep. Purif. Technol. 2018, 191, 424–432. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, L.; Liu, M.; Shu, Y.; Li, Q.; Wang, Y.; Qiu, D. Effect of the electronegativity on the electrosorption selectivity of anions during capacitive deionization. Chemosphere 2018, 195, 282–290. [Google Scholar] [CrossRef]
- Park, K.H.; Kwak, D.H. Electrosorption and electrochemical properties of activated-carbon sheet electrode for capacitive deionization. J. Electroanal. Chem. 2014, 732, 66–73. [Google Scholar] [CrossRef]
- Zhang, G.; He, Z.; Xu, W. A low-cost and high efficient zirconium-modified-Na-attapulgite adsorbent for fluoride removal from aqueous solutions. Chem. Eng. J. 2012, 183, 315–324. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, W.; Chen, L.; Chang, M.; Lee, C.; Tai, N. Electro-assisted selective uptake/release of phosphate using a graphene oxide/MgMn-layered double hydroxide composite. J. Mater. Chem. A 2019, 7, 3962–3970. [Google Scholar] [CrossRef]
- Yang, K.; Ying, T.; Yiacoumi, S.; Tsouris, A.C.; Vittoratos, E. Electrosorption of ions from aqueous solutions by carbon aerogel: An electrical double layer model. Langmuir 2001, 17, 1961–1969. [Google Scholar] [CrossRef]
- Sun, Z.; Li, Q.; Chai, L.; Shu, Y.; Wang, Y.; Qiu, D. Effect of the chemical bond on the electrosorption and desorption of anions during capacitive deionization. Chemosphere 2019, 229, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Biesheuvel, P.M.; van Limpt, B.; van der Wal, A. Dynamic adsorption/desorption process model for capacitive deionization. J. Phys. Chem. C 2009, 113, 5636–5640. [Google Scholar] [CrossRef]
- Xing, W.; Liang, J.; Tang, W.; Zeng, G.; Wang, X.; Li, X.; Jiang, L.; Luo, Y.; Li, X.; Tang, N.; et al. Perchlorate removal from brackish water by capacitive deionization: Experimental and theoretical investigations. Chem. Eng. J. 2019, 361, 209–218. [Google Scholar] [CrossRef]
- He, D.; Chi, E.W.; Tang, W.; Kovalsky, P.; Waite, T.D. Faradaic reactions in water desalination by batch-mode Capacitive Deionization. Environ. Sci. Technol. Lett. 2016, 3, 222–226. [Google Scholar] [CrossRef]
- Comninellis, C.; Nerini, A. Anodic oxidation of phenol in the presence of NaCl for wastewater treatment. J. Appl. Electrochem. 1995, 25, 23–28. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Rodrigo, M.A.; Sirés, I.; Scialdone, O. Single and coupled electrochemical processes and reactors for the abatement of organic water pollutants: A critical review. Chem. Rev. 2015, 115, 13362–13407. [Google Scholar] [CrossRef]
- Jung, Y.J.; Baek, K.W.; Oh, B.S.; Kang, J.-W. An investigation of the formation of chlorate and perchlorate during electrolysis using Pt/Ti electrodes: The effects of pH and reactive oxygen species and the results of kinetic studies. Water Res. 2010, 44, 5345–5355. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, J.M. Mechanistic insights for the electro-Fenton regeneration of carbon materials saturated with methyl orange: Dominance of electrodesorption. J. Hazard. Mater. 2019, 367, 59–67. [Google Scholar] [CrossRef]
- Lee, D.H.; Ryu, T.; Shin, J.; Ryu, J.C.; Chung, K.S.; Kim, Y.H. Selective lithium recovery from aqueous solution using a modified membrane capacitive deionization system. Hydrometallurgy 2017, 173, 283–288. [Google Scholar] [CrossRef]
- None, Lead oxidation work may lead to standards. Chem. Eng. News 1966, 44, 51.
- Xiong, W.; Tong, J.; Yang, Z.; Zeng, G.; Zhou, Y.; Wang, D.; Song, P.; Xu, R.; Zhang, C.; Cheng, M. Adsorption of phosphate from aqueous solution using iron-zirconium modified activated carbon nanofiber: Performance and mechanism. J. Colloid Interface Sci. 2017, 493, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Du, J.; Zhang, J.; David, W.; Liu, P.; Faheem, M.; Zhu, X.; Yang, J.; Bao, J. Microbially-mediated synthesis of activated carbon derived from cottonseed husks for enhanced sulfanilamide removal. J. Hazard. Mater. 2022, 426, 127811. [Google Scholar] [CrossRef]
- Yang, K.L.; Yiacoumi, S.; Tsouris, C. Proton adsorption and electrical double-layer formation inside charged platinum nanochannels. Nano Lett. 2004, 2, 1433–1437. [Google Scholar] [CrossRef]
- Ying, T.Y.; Yang, K.L.; Yiacoumi, S.; Tsouris, C. Electrosorption of ions from aqueous solutions by nanostructured carbon aerogel. J. Colloid Interface Sci. 2002, 250, 18–27. [Google Scholar] [CrossRef]
- Bo, S.; Luo, J.; An, Q.; Xiao, Z.; Wang, H.; Cai, W.; Zhai, S.; Li, Z. Efficiently selective adsorption of Pb(II) with functionalized alginate-based adsorbent in batch/column systems: Mechanism and application simulation. J. Clean. Prod. 2020, 250, 119585. [Google Scholar] [CrossRef]
- Han, L.; Karthikeyan, K.G.; Anderson, M.A.; Wouters, J.J.; Gregory, K.B. Mechanistic insights into the use of oxide nanoparticles coated asymmetric electrodes for capacitive deionization. Electrochim. Acta 2013, 90, 573–581. [Google Scholar] [CrossRef]
- Godino, M.P.; Barragán, V.M.; Izquierdo, M.A.; Villaluenga, J.P.G.; Seoane, B.; Ruiz-Bauzá, C. Study of the activation energy for transport of water and methanol through a Nafion membrane. Chem. Eng. J. 2009, 152, 20–25. [Google Scholar] [CrossRef]
- Qu, Y.; Campbell, P.G.; Hemmatifar, A.; Knipe, J.M.; Loeb, C.K.; Reidy, J.J.; Hubert, M.A.; Stadermann, M.; Santiago, G.J. Charging and Transport Dynamics of a Flow-Through Electrode Capacitive Deionization System. J Phys. Chem. B 2018, 122, 240–249. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Anode | Cathode | Sign | Cathode Potential/V |
|---|---|---|---|---|
| 1 | Titanium (Ti) | Titanium (Ti) | Ti-Ti | −0.22 |
| 2 | Graphite (G) | Titanium (Ti) | G-Ti | −1.70 |
| 3 | Graphite (G) | Lead (Pb) | G-Pb | −0.88 |
| 4 | Graphite (G) | Graphite (G) | G-G | −0.71 |
| 5 | Graphite (G) | Copper (Cu) | G-Cu | −1.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, J.; Wang, H.; Jin, R.; Liu, P.; Li, Y.; Wang, Y.; Wang, Q.; Sun, Z. Enhanced Electrodesorption Performance via Cathode Potential Extension during Capacitive Deionization. Appl. Sci. 2022, 12, 2874. https://doi.org/10.3390/app12062874
Fu J, Wang H, Jin R, Liu P, Li Y, Wang Y, Wang Q, Sun Z. Enhanced Electrodesorption Performance via Cathode Potential Extension during Capacitive Deionization. Applied Sciences. 2022; 12(6):2874. https://doi.org/10.3390/app12062874
Chicago/Turabian StyleFu, Jie, Haifang Wang, Riya Jin, Pengxiao Liu, Ying Li, Yunyan Wang, Qingwei Wang, and Zhumei Sun. 2022. "Enhanced Electrodesorption Performance via Cathode Potential Extension during Capacitive Deionization" Applied Sciences 12, no. 6: 2874. https://doi.org/10.3390/app12062874
APA StyleFu, J., Wang, H., Jin, R., Liu, P., Li, Y., Wang, Y., Wang, Q., & Sun, Z. (2022). Enhanced Electrodesorption Performance via Cathode Potential Extension during Capacitive Deionization. Applied Sciences, 12(6), 2874. https://doi.org/10.3390/app12062874