
Ambient Melting Behavior of Stoichiometric Uranium-Plutonium Mixed Oxide Fuel †



Abstract
:1. Introduction
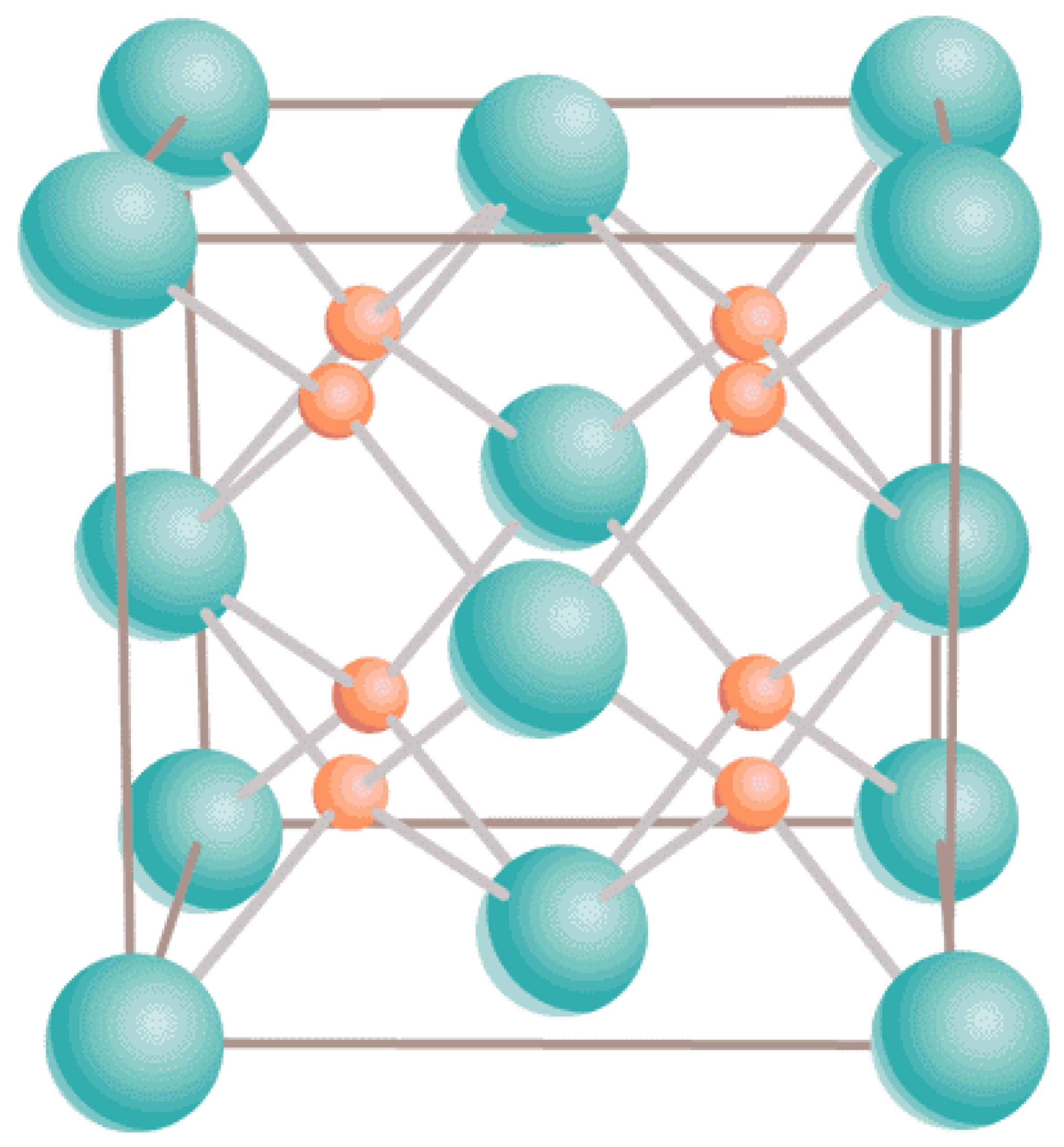
1.1. Mixed Oxide Fuel (MOX)
1.2. Ambient Melting Behavior
2. Methods
2.1. LDA + U Methodology
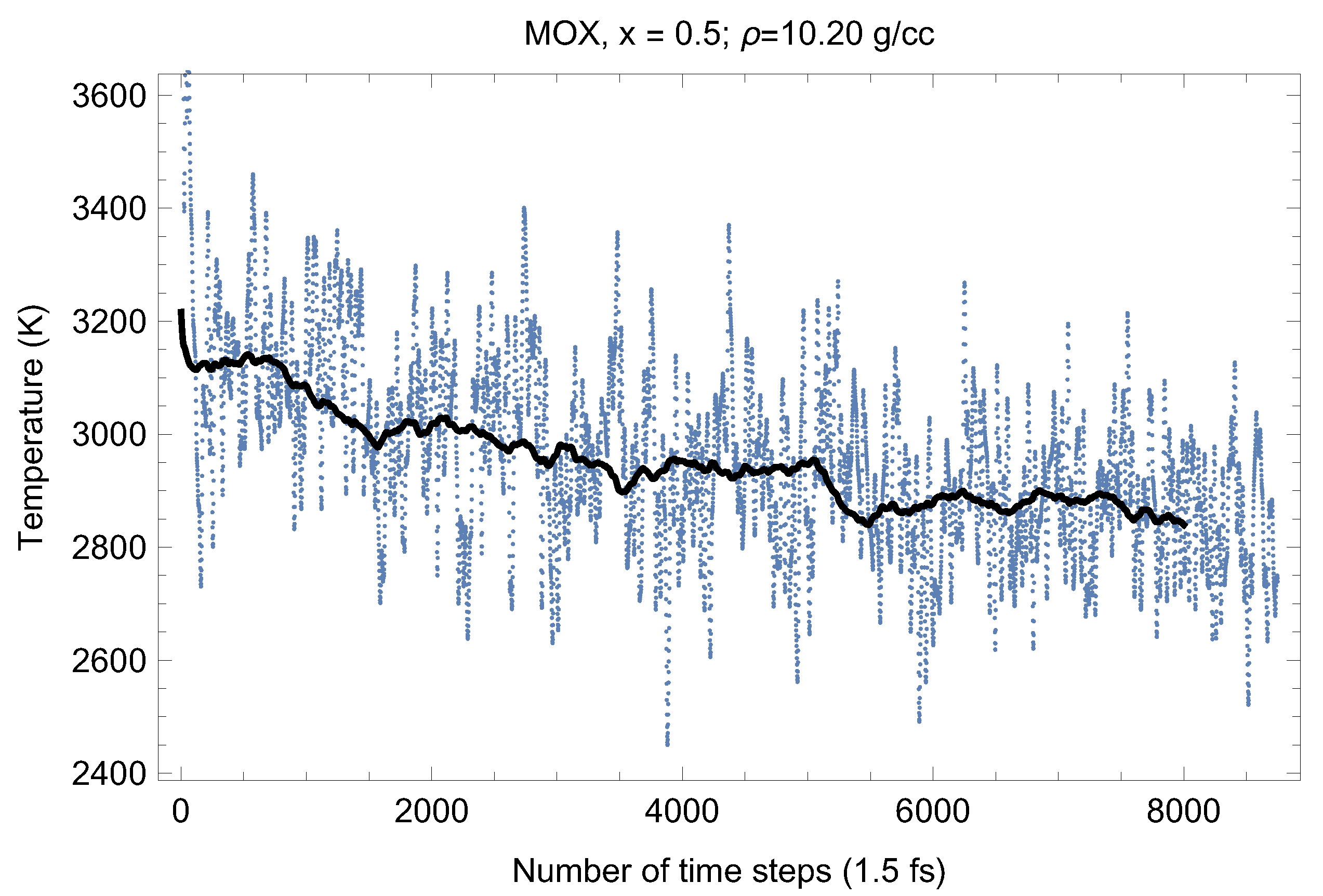
2.2. Melting Simulations
3. Results








4. Discussion
4.1. Energy Convergence of Our Results with Respect to the System Size




4.2. Initial Slope of the Melting Curve of PuO2
4.3. The Ambient Phase Diagram of Stoichiometric MOX
4.4. Potential Ambient Density–Melting Point Systematics of MOX
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shellenberger, M. Had They Bet on Nuclear, Not Renewables, Germany & California Would Already Have 100% Clean Power. Available online: https://www.forbes.com/sites/michaelshellenberger/2018/09/11/had-they-bet-on-nuclear-not-renewables-germany-california-would-already-have-100-clean-power/#e9a4e94e0d44 (accessed on 2 May 2023).
- Carbajo, J.J.; Yoder, G.L.; Popov, S.G.; Ivanov, V.K. A review of the thermophysical properties of MOX and UO2 fuels. J. Nucl. Mater. 2001, 299, 181–198. [Google Scholar] [CrossRef]
- Guéneau, C.; Chatillon, C.; Sundman, B. Thermodynamic modelling of the plutonium–oxygen system. J. Nucl. Mater. 2008, 378, 257–272. [Google Scholar] [CrossRef]
- Kato, M.; Morimoto, K.; Sugata, H.; Konashi, K.; Kashimura, M.; Abe, T. Solidus and liquidus of plutonium and uranium mixed oxide. J. Alloys Compd. 2008, 452, 48–53. [Google Scholar] [CrossRef]
- Kato, M.; Morimoto, K.; Sugata, H.; Konashi, K.; Kashimura, M.; Abe, T. Solidus and liquidus temperatures in the UO2–PuO2 system. J. Nucl. Mater. 2008, 373, 237–245. [Google Scholar] [CrossRef]
- De Bruycker, F.; Boboridis, K.; Manara, D.; Pöml, P.; Rini, M.; Konings, R.J. Reassessing the melting temperature of PuO2. Mater. Today 2010, 13, 52–55. [Google Scholar] [CrossRef]
- Böhler, R.; Well, M.J.; Prieur, D.; Cakir, P.; Vitova, T.; Pruessmann, T.; Pidchenko, I.; Hennig, C.; Guéneau, C.; Konings, R.J.; et al. Recent advances in the study of the UO2-PuO2 phase diagram at high temperatures. J. Nucl. Mater. 2014, 448, 330. [Google Scholar] [CrossRef]
- Fouquet-Métivier, P.; Martin, P.M.; Manara, D.; Dardenne, K.; Rothe, J.; Fossati, P.C.; Guéneau, C. Investigation of the solid/liquid phase transitions in the U–Pu–O system. Calphad 2023, 80, 102523-1–102523-18. [Google Scholar] [CrossRef]
- Errandonea, D.; MacLeod, S.G.; Ruiz-Fuertes, J.; Burakovsky, L.; McMahon, M.I.; Wilson, C.W.; Ibañez, J.; Daisenberger, D.; Popescu, C. High-pressure/high-temperature phase diagram of zinc. J. Phys. Condens. Matter. 2018, 30, 295402-1–295402-8. [Google Scholar] [CrossRef]
- Anzellini, S.; Monteseguro, V.; Bandiello, E.; Dewaele, A.; Burakovsky, L.; Errandonea, D. In situ characterization of the high pressure–high temperature melting curve of platinum. Sci. Rep. 2019, 9, 13034. [Google Scholar] [CrossRef]
- Burakovsky, L.; Burakovsky, N.; Preston, D.L. Ab initio melting curve of osmium. Phys. Rev. B 2015, 92, 174105. [Google Scholar] [CrossRef]
- Patel, N.N.; Sunder, M. High pressure melting curve of osmium up to 35 GPa. J. Appl. Phys. 2018, 125, 055902-1–055902-5. [Google Scholar] [CrossRef]
- Baty, S.R.; Burakovsky, L.; Errandonea, D. Ab initio phase diagram of copper. Crystals 2021, 11, 537. [Google Scholar] [CrossRef]
- Baty, S.R.; Burakovsky, L.; Errandonea, D. Ab initio phase diagram of silver. J. Phys. Condens. Matter. 2021, 33, 485901-1–485901-10. [Google Scholar] [CrossRef]
- Sims, M.; Briggs, R.; Volz, T.J.; Singh, S.; Hamel, S.; Coleman, A.L.; Coppari, F.; Erskine, D.J.; Gorman, M.G.; Sadigh, B.; et al. Experimental and theoretical examination of shock-compressed copper through the fcc to bcc to melt phase transitions. J. Appl. Phys. 2022, 132, 075902-1–075902-11. [Google Scholar] [CrossRef]
- Coleman, A.L.; Singh, S.; Vennari, C.E.; Smith, R.F.; Volz, T.J.; Gorman, M.G.; Clarke, S.M.; Eggert, J.H.; Coppari, F.; Fratanduono, D.E.; et al. Quantitative measurements of density in shock-compressed silver up to 330 GPa using x-ray diffraction. J. Appl. Phys. 2022, 131, 015901-1–015901-10. [Google Scholar] [CrossRef]
- Nakamura, H.; Machida, M.; Kato, M. LDA+U study on plutonium dioxide with spin–orbit couplings. Prog. Nucl. Sci. Technol. 2011, 2, 16. [Google Scholar] [CrossRef]
- Kvashnina, K.O.; Butorin, S.M.; Martin, P.; Glatzel, P. Chemical state of complex uranium oxides. Phys. Rev. Lett. 2013, 111, 235002–235005. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, B.; Zhang, P. Electronic and mechanical properties of ordered (Pu, U) O2 compounds: A density functional theory +U study. J. Nucl. Mater. 2013, 433, 345. [Google Scholar] [CrossRef]
- Allen, J.P.; Watson, G.W. Occupation matrix control of d- and f-electron localisations using DFT + U. Phys. Chem. Chem. Phys. 2014, 16, 21016–21031. [Google Scholar] [CrossRef]
- Chen, J.-L.; Kaltsoyannis, N. DFT + U Study of uranium dioxide and plutonium dioxide with occupation matrix control. J. Phys. Chem. C 2022, 126, 11426–11435. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.C.; Holmes, N.R. A mechanism for the UO2 to α-U3O8 phase transformation. J. Nucl. Mater. 1995, 223, 231. [Google Scholar] [CrossRef]
- Wan, M.; Zhang, L.; Du, J.; Huang, D.; Wang, L.; Jiang, G. The MD simulation of thermal properties of plutonium dioxide. Phys. B 2012, 407, 4595. [Google Scholar]
- Dworkin, A.S.; Bredig, M.A. Diffuse transition and melting in fluorite and antifluorite type of compounds. Heat content of potassium sulfide from 298 to 1260. degree. K. J. Phys. Chem. 1968, 72, 1277. [Google Scholar] [CrossRef]
- Chroneos, A.; Fitzpatrick, M.E.; Tsoukalas, L.H. Describing oxygen self-diffusion in PuO2 by connecting point defect parameters with bulk properties. J. Mater. Sci. Mater. Electron. 2015, 26, 3287. [Google Scholar] [CrossRef]
- Günay, S.D.; Akgenç, B.; Taşseven, Ç. Modeling Superionic Behavior of Plutonium Dioxide. High Temp. Mater. Proc. 2016, 35, 999. [Google Scholar] [CrossRef]
- Manara, D.; Ronchi, C.; Sheindlin, M.; Lewis, M.; Brykin, M. Manara D, Ronchi C, Sheindlin M, Lewis M, Brykin M. Melting of stoichiometric and hyperstoichiometric uranium dioxide. J. Nucl. Mater. 2005, 342, 148. [Google Scholar] [CrossRef]
- Lyon, W.L.; Baily, W.E. The solid–liquid phase diagram for the UO2-PuO2 system. J. Nucl. Mater. 1967, 22, 332–339. [Google Scholar] [CrossRef]
- De Bruycker, F.; Boboridis, K.; Konings, R.J.; Rini, M.; Eloirdi, R.; Guéneau, C.; Dupin, N.; Manara, D. On the melting behaviour of uranium/plutonium mixed dioxides with high-Pu content: A laser heating study. J. Nucl. Mater. 2011, 419, 186–193. [Google Scholar] [CrossRef]
- Adamson, A.M.G.; Aitken, E.A.; Caputi, R.W. Experimental and thermodynamic evaluation of the melting behavior of irradiated oxide fuels. J. Nucl. Mater. 1985, 130, 349–365. [Google Scholar] [CrossRef]
- Konno, K.; Hirosawa, T. Melting temperature of mixed oxide fuels for fast reactors. J. Nucl. Sci. Technol. 2002, 39, 771–777. [Google Scholar] [CrossRef]
- Burakovsky, L.; Preston, D.L.; Silbar, R.R. Melting as a dislocation-mediated phase transition. Phys. Rev. B 2000, 61, 15011. [Google Scholar] [CrossRef]
- Burakovsky, L.; Preston, D.L.; Silbar, R.R. Analysis of dislocation mechanism for melting of elements: Pressure dependence. J. Appl. Phys. 2000, 88, 6294–6301. [Google Scholar] [CrossRef]
- Preston, D.L.; Wallace, D.C. A model of the shear modulus. Solid state communications. Solid State Commun. 1992, 81, 277–281. [Google Scholar] [CrossRef]
- Strach, M.; Manara, D.; Belin, R.C.; Rogez, J. Melting behavior of mixed U–Pu oxides under oxidizing conditions. Nucl. Instrum. Methods Phys. Res. B 2016, 374, 125–128. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | (K) | (K) | Lattice Constant (Å) | Density (g/cm) |
|---|---|---|---|---|
| 0 | 3150 | 135 | 5.700 | 9.685 |
| 0.25 | 2910 | 135 | 5.653 | 9.984 |
| 0.5 | 2870 | 135 | 5.624 | 10.20 |
| 0.7 | 2670 | 135 | 5.585 | 10.46 |
| 0.9 | 2750 | 135 | 5.572 | 10.58 |
| 1 | 3050 | 135 | 5.590 | 10.50 |
| Lattice Constant (Å) | Density (g/cm) | (GPa) | (K) | (K) |
|---|---|---|---|---|
| 5.597 | 10.46 | −1.8 | 2780 | 125 |
| 5.590 | 10.50 | −0.6 | 3010 | 125 |
| 5.587 | 10.51 | 0.8 | 3090 | 125 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burakovsky, L.; Ramsey, S.D.; Baty, R.S. Ambient Melting Behavior of Stoichiometric Uranium-Plutonium Mixed Oxide Fuel. Appl. Sci. 2023, 13, 6303. https://doi.org/10.3390/app13106303
Burakovsky L, Ramsey SD, Baty RS. Ambient Melting Behavior of Stoichiometric Uranium-Plutonium Mixed Oxide Fuel. Applied Sciences. 2023; 13(10):6303. https://doi.org/10.3390/app13106303
Chicago/Turabian StyleBurakovsky, Leonid, Scott D. Ramsey, and Roy S. Baty. 2023. "Ambient Melting Behavior of Stoichiometric Uranium-Plutonium Mixed Oxide Fuel" Applied Sciences 13, no. 10: 6303. https://doi.org/10.3390/app13106303
APA StyleBurakovsky, L., Ramsey, S. D., & Baty, R. S. (2023). Ambient Melting Behavior of Stoichiometric Uranium-Plutonium Mixed Oxide Fuel. Applied Sciences, 13(10), 6303. https://doi.org/10.3390/app13106303