
COX2-Inhibitory and Cytotoxic Activities of Phytoconstituents of Matricaria chamomilla L.



Abstract
:1. Introduction
2. Materials and Methods
2.1. Phytochemical Analysis
Chemicals
2.2. Instrumentation and Chromatographic Conditions of Liquid Chromatography with a Diode Array Detector–Quadrupole Time-of-Flight Mass Spectrometry (LC–DAD–QToF)
2.3. Virtual Drug Screening and Molecular Docking
2.4. Fluorometric COX2 Inhibitor Screening Assay
2.5. Pharmacological Testing and Gene/Protein Expression Profiling of Tumor Cell Lines
2.6. Growth Inhibition Assay
2.7. Kaplan–Meier Survival Analysis
2.8. Immunofluorescence Microscopy of GFP Tagged α-Tubulin
3. Results
3.1. Phytochemical Analysis
3.2. Cytotoxicity Assay
3.3. Molecular Docking In Silico
3.4. Inhibition of COX2 Enzyme Activity In Vitro
3.5. Cytotoxicity against Tumor Cells In Vitro
3.6. Oncobiogram Analysis
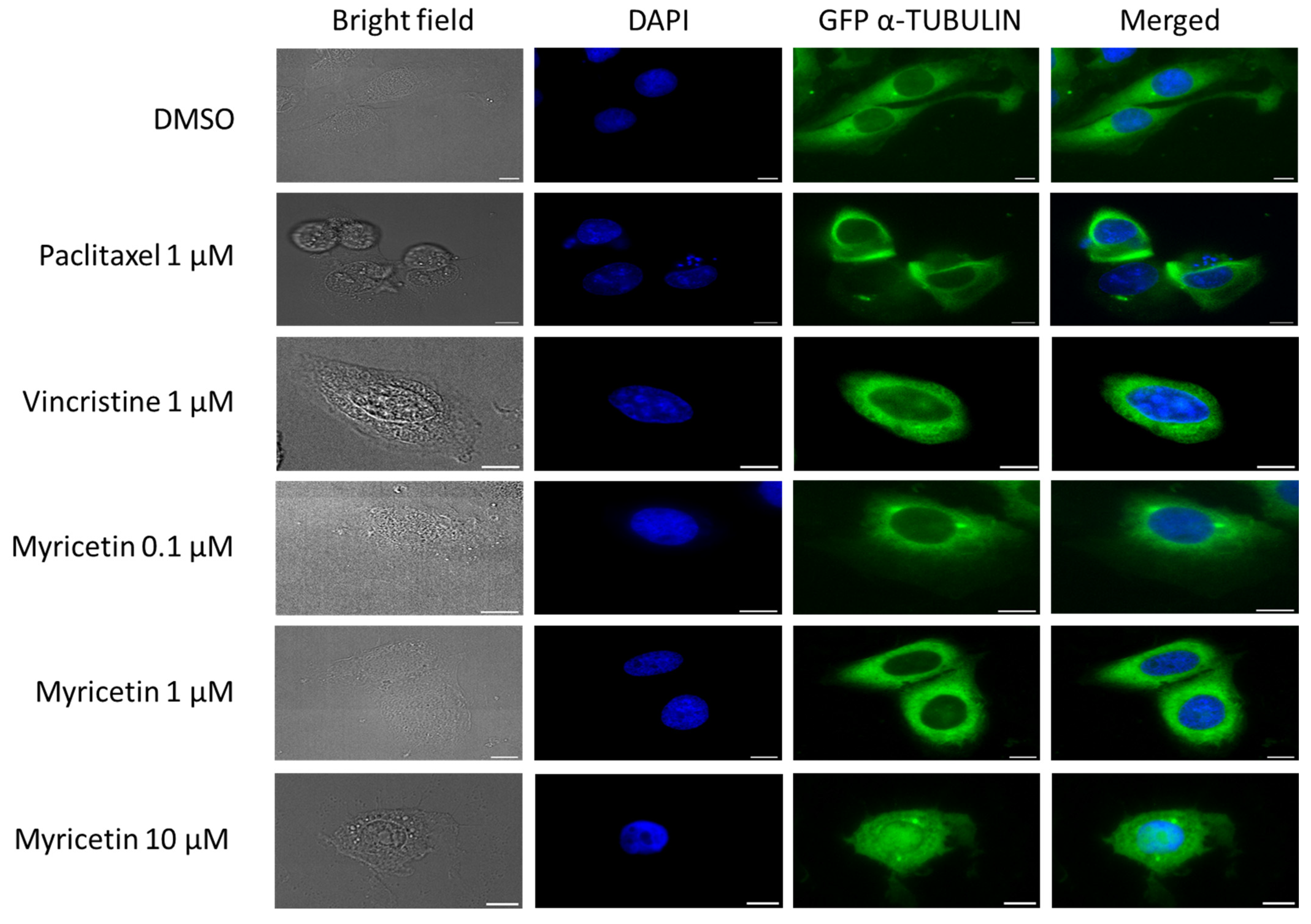
3.7. Effect of Myricetin on α-Tubulin
3.8. Proteome Analysis
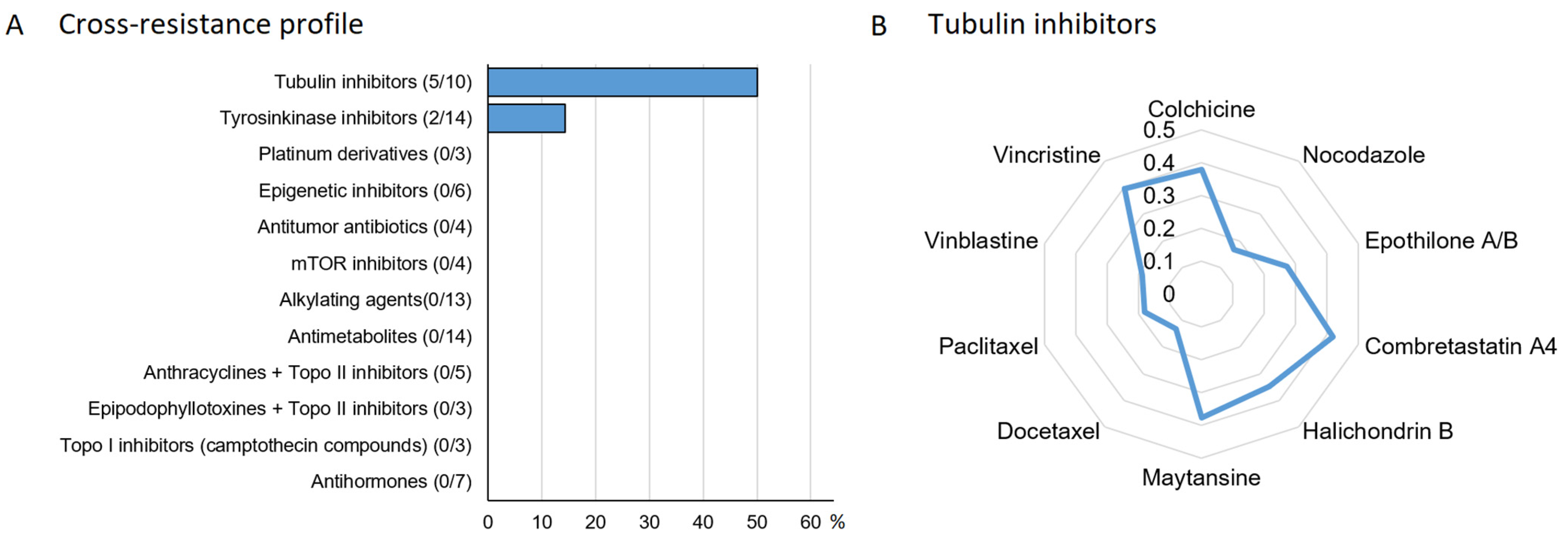
3.9. Drug Resistance Profiling of Myricetin
3.10. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKay, D.L.; Blumberg, J.B. The Role of Tea in Human Health: An Update. J. Am. Coll. Nutr. 2013, 21, 1–13. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Shankar, E.; Gupta, S. Chamomile: A Herbal Medicine of the Past with a Bright Future (Review). Mol. Med. Rep. 2010, 3, 895–901. [Google Scholar]
- Hassan, D.M.A.; Salah-Eldin, A.A. Amerolative Influence of Chamomile (Matricaria recutita L.) on Synthetic Food Additive Induced Probable Toxicity in Male Albino Rats. J. Food Dairy Sci. J. 2021, 12, 161–170. [Google Scholar] [CrossRef]
- CFR—Code of Federal Regulations Title 21; Food and Drug Administration: Silver Spring, MD, USA, 2023; p. 3.
- Sándor, Z.; Mottaghipisheh, J.; Veres, K.; Hohmann, J.; Bencsik, T.; Horváth, A.; Kelemen, D.; Papp, R.; Barthó, L.; Csupor, D. Evidence Supports Tradition: The in Vitro Effects of Roman Chamomile on Smooth Muscles. Front. Pharmacol. 2018, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Pirouzpanah, S.; Mahboob, S.; Sanayei, M.; Hajaliloo, M.; Safaeiyan, A. The Effect of Chamomile Tea Consumption on Inflammation among Rheumatoid Arthritis Patients: Randomized Clinical Trial. Prog. Nutr. 2017, 19, 27–33. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Dmytriv, T.R.; Melnychuk, A.V.; Strilets, N.V.; Storey, K.B.; Lushchak, V.I. Chamomile as a Potential Remedy for Obesity and Metabolic Syndrome. EXCLI J. 2021, 20, 1261–1286. [Google Scholar] [CrossRef]
- Srivastava, J. Antiproliferative and Apoptotic Effects of Chamomile Extract in Various Human Cancer Cells. J. Agric. Food Chem. 2007, 55, 9470–9478. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Pandey, M.; Gupta, S. Chamomile, a Novel and Selective COX-2 Inhibitor with Anti-Inflammatory Activity. Life Sci. 2009, 85, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.M.; Chen, C.H. Effects of an Intervention with Drinking Chamomile Tea on Sleep Quality and Depression in Sleep Disturbed Postnatal Women: A Randomized Controlled Trial. J. Adv. Nurs. 2016, 72, 306–315. [Google Scholar] [CrossRef]
- Gh, D.; Kermanian, S.; Mozaffari-Khosravi, H.; Dastgerdi, G.; Zavar-Reza, J.; Rahmanian, M. The Effect of Chamomile Tea versus Black Tea on Glycemic Control and Blood Lipid Profiles in Depressed Patients with Type 2 Diabetes: A Randomized Clinical Trial. J. Nutr. Food Secur. 2018, 3, 157–166. [Google Scholar]
- Ghamchini, V.M.; Salami, M.; Mohammadi, G.R.; Moradi, Z.; Kavosi, A.; Movahedi, A.; Bidkhori, M.; Aryaeefar, M.R. The Effect of Chamomile Tea on Anxiety and Depression in Cancer Patients Treated with Chemotherapy. J. Young Pharm. 2019, 11, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Paula Gardiner Chamomile (Matricaria Recutita, Anthemis Nobilis). The Longwood Herbal Task Force and The Center for Holistic Pediatric Education and Research. 1999, pp. 1–21. Available online: https://tratamientocelular.com/papers/cmran.pdf (accessed on 31 March 2023).
- El Joumaa, M.M.; Borjac, J.M. Matricaria Chamomilla: A Valuable Insight into Recent Advances in Medicinal Uses and Pharmacological Activities. Phytochem. Rev. 2022, 21, 1913–1940. [Google Scholar] [CrossRef]
- Wu, B.Y.; Liu, C.T.; Su, Y.L.; Chen, S.Y.; Chen, Y.H.; Tsai, M.Y. A Review of Complementary Therapies with Medicinal Plants for Chemotherapy-Induced Peripheral Neuropathy. Complement. Ther. Med. 2019, 42, 226–232. [Google Scholar] [CrossRef]
- Salehi, B.; Jornet, P.L.; López, E.P.F.; Calina, D.; Sharifi-Rad, M.; Ramírez-Alarcón, K.; Forman, K.; Fernández, M.; Martorell, M.; Setzer, W.N.; et al. Plant-Derived Bioactives in Oral Mucosal Lesions: A Key Emphasis to Curcumin, Lycopene, Chamomile, Aloe Vera, Green Tea and Coffee Properties. Biomolecules 2019, 9, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Ling, C.; Wang, C.; Wang, H.; Su, L.; Yang, L.; Jiang, W.; Yu, X.; Zheng, L.; Feng, Z.; et al. Analysis of Terpenoid Biosynthesis Pathways in German Chamomile (Matricaria recutita) and Roman Chamomile (Chamaemelum nobile) Based on Co-Expression Networks. Genomics 2020, 112, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Nováková, L.; Vildová, A.; Mateus, J.P.; Gonalves, T.; Solich, P. Development and Application of UHPLC-MS/MS Method for the Determination of Phenolic Compounds in Chamomile Flowers and Chamomile Tea Extracts. Talanta 2010, 82, 1271–1280. [Google Scholar] [CrossRef]
- Power, F.B.; Browning, H. CCXI.—The Constituents of the Flowers of Matricaria Chamomilla. J. Chem. Soc. Trans. 1914, 105, 2280–2291. [Google Scholar] [CrossRef] [Green Version]
- Lefort, É.C.; Blay, J. Apigenin and Its Impact on Gastrointestinal Cancers. Mol. Nutr. Food Res. 2013, 57, 126–144. [Google Scholar] [CrossRef]
- Xu, Y.; Xin, Y.; Diao, Y.; Lu, C.; Fu, J.; Luo, L.; Yin, Z. Synergistic Effects of Apigenin and Paclitaxel on Apoptosis of Cancer Cells. PLoS ONE 2011, 6, e29169. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Shukla, S.; Gupta, S. Apigenin and Cancer Chemoprevention: Progress, Potential and Promise (Review). Int. J. Oncol. 2007, 30, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, J.K.; Gupta, S. Extraction, Characterization, Stability and Biological Activity of Flavonoids Isolated from Cham-omile Flowers. Mol. Cell. Pharmacol. 2009, 1, 138. [Google Scholar] [CrossRef]
- Bhaskaran, N.; Shukla, S.; Srivastava, J.K.; Gupta, S. Chamomile: An Anti-Inflammatory Agent Inhibits Inducible Nitric Oxide Synthase Expression by Blocking RelA/P65 Activity. Int. J. Mol. Med. 2010, 26, 935–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakariar, R.; Yaqoob, M.M.; Gilroy, D.W. COX-2 in Inflammation and Resolution. Mol. Interv. 2006, 6, 199. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.S.; Mann, M.; DuBois, R.N. The Role of Cyclooxygenases in Inflammation, Cancer, and Development. Oncogene 2000, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardeh, D.; Wang, D.; Costigan, M.; Lazarus, M.; Saper, C.B.; Woolf, C.J.; Fitzgerald, G.A.; Samad, T.A. COX2 in CNS Neural Cells Mediates Mechanical Inflammatory Pain Hypersensitivity in Mice. J. Clin. Investig. 2009, 119, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Seibert, K.; Masferrer, J.L. Role of Inducible Cyclooxygenase (COX-2) in Inflammation. Receptor 1994, 4, 17–23. [Google Scholar]
- Avula, B.; Bae, J.Y.; Wang, Y.H.; Wang, M.; Ali, Z.; Khan, I.A. Chemical Profiling and Characterization of Anthraquinones from Two Bulbine Species and Dietary Supplements Using Liquid Chromatography–High Resolution Mass Spectrometry. J. AOAC Int. 2021, 104, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Avula, B.; Sagi, S.; Masoodi, M.H.; Bae, J.Y.; Wali, A.F.; Khan, I.A. Quantification and Characterization of Phenolic Compounds from Northern Indian Propolis Extracts and Dietary Supplements. J. AOAC Int. 2020, 103, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Supianto, A.A.; Nurdiansyah, R.; Weng, C.W.; Zilvan, V.; Yuwana, R.S.; Arisal, A.; Pardede, H.F.; Lee, M.M.; Huang, C.H.; Ng, K.L. Cluster-Based Text Mining for Extracting Drug Candidates for the Prevention of COVID-19 from the Biomedical Literature. J. Taibah Univ. Med. Sci. 2023, 18, 787–801. [Google Scholar] [CrossRef]
- PyRx (Version 0.8). Available online: https://pyrx.sourceforge.io/ (accessed on 31 March 2023).
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry HHS Public Access. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef] [Green Version]
- RCSB PDB—5F1A: The Crystal Structure of Salicylate Bound to Human Cyclooxygenase-2. Available online: https://www.rcsb.org/structure/5F1A (accessed on 31 March 2023).
- UCSF Chimera Home Page. Available online: https://www.cgl.ucsf.edu/chimera/ (accessed on 30 March 2023).
- Search|Scripps Research. Available online: https://www.scripps.edu/search/?s=autodocktools (accessed on 31 March 2023).
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Huey, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. User Guide AutoDock Version 4.2 Updated for Version 4.2.6 Automated Docking of Flexible Ligands to Flexible Receptors; 1991. Available online: http://autodock.scripps.edu/ (accessed on 30 March 2023).
- AutoDock. Available online: https://autodock.scripps.edu/ (accessed on 31 March 2023).
- Yadav, T.C.; Kumar, N.; Raj, U.; Goel, N.; Vardawaj, P.K.; Prasad, R.; Pruthi, V. Exploration of Interaction Mechanism of Tyrosol as a Potent Anti-Inflammatory Agent. J. Biomol. Struct. Dyn. 2019, 38, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Free Download: BIOVIA Discovery Studio Visualizer—Dassault Systèmes. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 30 March 2023).
- ab283401; COX-2 Inhibitor Screening Kit (Fluorometric). Abcam plc.: Singapore, 2020.
- About NCI—NCI. Available online: https://www.cancer.gov/about-nci (accessed on 30 March 2023).
- Alley, M.C.; Scudiere, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay1. Cancer Res. 1988, 48, 89–90. [Google Scholar]
- Developmental Therapeutics Program (DTP). Available online: https://dtp.cancer.gov/ (accessed on 30 March 2023).
- Investigations of Anticancer Compounds from Camellia Sinensis: Green, Black, and White Tea—ProQuest. Available online: https://www.proquest.com/docview/1625976354?fromopenview=true&pq-origsite=gscholar (accessed on 22 July 2023).
- Kuete, V.; Wabo, H.K.; Eyong, K.O.; Feussi, M.T.; Wiench, B.; Krusche, B.; Tane, P.; Folefoc, G.N.; Efferth, T. Anticancer Activities of Six Selected Natural Compounds of Some Cameroonian Medicinal Plants. PLoS ONE 2011, 6, e21762. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Meier Plotter. Available online: https://kmplot.com/analysis/ (accessed on 30 March 2023).
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhou, M.; Boulos, J.C.; Klauck, S.M.; Efferth, T.; Zhou, M.; Boulos, J.C.; Efferth, T.; Klauck, S.M. The Cardiac Glycoside ZINC253504760 Induces Parthanatos-Type Cell Death and G2/M Arrest via Downregulation of MEK1/2 Phosphorylation in Leukemia Cells. Cell Biol. Toxicol. 2023, 3, 1–27. [Google Scholar] [CrossRef]
- Vezzani, B.; Carinci, M.; Previati, M.; Giacovazzi, S.; Della Sala, M.; Gafà, R.; Lanza, G.; Wieckowski, M.R.; Pinton, P.; Giorgi, C. Epigenetic Regulation: A Link between Inflammation and Carcinogenesis. Cancers 2022, 14, 1221. [Google Scholar] [CrossRef]
- Saeed, M.; Efferth, T.; Kadioglu, O.; Khalid, H.; Sugimoto, Y. Activity of the Dietary Flavonoid, Apigenin, against Multidrug-Resistant Tumor Cells as Determined by Pharmacogenomics and Molecular Docking. J. Immunother. Cancer 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.-F.; Luo, M.; Zhao, C.-J.; Li, C.-Y.; Gu, C.-B.; Wang, W.; Zu, Y.-G.; Efferth, T.; Fu, Y.-J. UV-Induced Changes of Active Components and Antioxidant Activity in Postharvest Pigeon Pea [Cajanus cajan (L.) Millsp.] Leaves. J. Agric. Food Chem. 2013, 61, 1165–1171. [Google Scholar] [CrossRef]
- Adham, A.N.; Abdelfatah, S.; Naqishbandi, A.M.; Mahmoud, N.; Efferth, T. Cytotoxicity of Apigenin toward Multiple Myeloma Cell Lines and Suppression of iNOS and COX-2 Expression in STAT1-Transfected HEK293 Cells. Phytomedicine 2021, 80, 153371. [Google Scholar] [CrossRef]
- Kuete, V.; Efferth, T. Molecular Determinants of Cancer Cell Sensitivity and Resistance towards the Sesquiterpene Farnesol. Pharmaceuticals 2011, 4, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T.; Koch, E. Complex Interactions between Phytochemicals. The Multi-Target Therapeutic Concept of Phytotherapy. Curr. Drug Targets 2011, 12, 122–132. [Google Scholar] [PubMed]
- Bhattacherjee, S.; Ray, A.; Chakraborty, K. Therapeutic and Ethnopharmacological Role of Chamomile (Matricaria chamomilla L.) and Its Holistic Impact on Genomics-A Comprehensive Review. World J. Pharm. Res. 2022, 11, 705. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Apigenin: A Promising Molecule for Cancer Prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Yasui, Y.; Tanaka, T. Protein Expression Analysis of Inflammation-Related Colon Carcinogenesis. J. Carcinog. 2009, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Singh, A. (Ed.) Herbalism, Phytochemistry and Ethnopharmacology; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Dai, Y.L.; Li, Y.; Wang, Q.; Niu, F.J.; Li, K.W.; Wang, Y.Y.; Wang, J.; Zhou, C.Z.; Gao, L.N. Chamomile: A Review of Its Traditional Uses, Chemical Constituents, Pharmacological Activities and Quality Control Studies. Molecules 2023, 28, 133. [Google Scholar] [CrossRef]
- Baek, G.H.; Yang, S.W.; Yun, C.I.; Lee, J.G.; Kim, Y.J. Determination of Methylxanthine Contents and Risk Characterisation for Various Types of Tea in Korea. Food Control 2022, 132, 108543. [Google Scholar] [CrossRef]
- Yoo, K.; Hwang, I.K.; Moon, B. Comparative Flavonoids Contents of Selected Herbs and Associations of Their Radical Scavenging Activity with Antiproliferative Actions in V79-4 Cells. J. Food Sci. 2009, 74, C419–C425. [Google Scholar] [CrossRef]
- Cleverdon, R.; Elhalaby, Y.; McAlpine, M.D.; Gittings, W.; Ward, W.E. Total Polyphenol Content and Antioxidant Capacity of Tea Bags: Comparison of Black, Green, Red Rooibos, Chamomile and Peppermint over Different Steep Times. Beverages 2018, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- McKay, D.L.; Blumberg, J.B.; Research Labora-tory, A.; Mayer, J. A Review of the Bioactivity and Potential Health Benefits of Chamomile Tea (Matricaria recutita L.). Phytother. Res. 2006, 20, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Rizeq, B.; Gupta, I.; Ilesanmi, J.; AlSafran, M.; Rahman, M.D.M.; Ouhtit, A. The Power of Phytochemicals Combination in Cancer Chemoprevention. J. Cancer 2020, 11, 4521–4533. [Google Scholar] [CrossRef] [PubMed]
- Tanos, V.; Brzezinski, A.; Drize, O.; Strauss, N.; Peretz, T. Synergistic Inhibitory Effects of Genistein and Tamoxifen on Human Dysplastic and Malignant Epithelial Breast Cells In Vitro. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 102, 188–194. [Google Scholar] [CrossRef]
- Hemalswarya, S.; Doble, M. Potential Synergism of Natural Products in the Treatment of Cancer. Phytother. Res. 2006, 20, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, D.; Goode, J. Novartis Foundation. Cancer and Inflammation; John Wiley & Sons: Chichester, UK, 2004; ISBN 047085510X. [Google Scholar]
- Alhouayek, M.; Muccioli, G.G. COX-2-Derived Endocannabinoid Metabolites as Novel Inflammatory Mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Non-Redundant Functions of Cyclooxygenases: Oxygenation of Endocannabinoids. J. Biol. Chem. 2008, 283, 8065–8069. [Google Scholar] [CrossRef] [Green Version]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid Oxygenation by Cyclooxygenases, Lipoxygenases, and Cytochromes P450: Cross-Talk between the Eicosanoid and Endocannabinoid Signaling Pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The Role of P53 in Cancer Drug Resistance and Targeted Chemotherapy. Oncotarget 2017, 8, 8921. [Google Scholar] [CrossRef] [Green Version]
- Mbaveng, A.T.; Damen, F.; Guefack, M.G.F.; Tankeo, S.B.; Abdelfatah, S.; Bitchagno, G.T.M.; Çelik, İ.; Kuete, V.; Efferth, T. 8,8-Bis-(Dihydroconiferyl)-Diferulate Displayed Impressive Cytotoxicity towards a Panel of Human and Animal Cancer Cells. Phytomedicine 2020, 70, 153215. [Google Scholar] [CrossRef]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-Regulation of Cyclooxygenase 2 Gene Expression in Human Colorectal Adenomas and Adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Yatabe, Y.; Achiwa, H.; Muramatsu, H.; Kozaki, K.-I.; Nakamura, S.; Ogawa, M.; Mitsudomi, T.; Sugiura, T.; Takahashi, T. Increased Expression of Cyclooxygenase 2 Occurs Frequently in Human Lung Cancers, Specifically in Adenocarcinomas. Cancer Res. 1998, 58, 1761–3764. [Google Scholar]
- Rizzo, M.T. Cyclooxygenase-2 in Oncogenesis. Clin. Chim. Acta 2011, 412, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Discovery and Ranking of the Most Robust Prognostic Biomarkers in Serous Ovarian Cancer. Geroscience 2023, submitted. [CrossRef]
- Banerjee, S.; Hwang, D.-J.; Li, W.; Miller, D.D. Molecules Current Advances of Tubulin Inhibitors in Nanoparticle Drug Delivery and Vascular Disruption/Angiogenesis. Molecules 2016, 21, 1468. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Evers, B.M.; Sheng, H. Prostaglandin E2 Synergistically Enhances Receptor Tyrosine Kinase-Dependent Signaling System in Colon Cancer Cells. J. Biol. Chem. 2004, 279, 14287–14293. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Michalopoulos, G.K.; Wu, T. Prostaglandin E2 Receptor EP1 Transactivates EGFR/MET Receptor Tyrosine Kinases and Enhances Invasiveness in Human Hepatocellular Carcinoma Cells. J. Cell Physiol. 2006, 207, 261–270. [Google Scholar] [CrossRef]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brüne, B. Macrophages Programmed by Apoptotic Cells Promote Angiogenesis via Prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, S.; Wang, Y.; Liu, X.; Zhou, L.; Yin, P.; Fan, Z.; Li, Q. COX-2 Contributes to P-Glycoprotein-Mediated Multidrug Resistance via Phosphorylation of c-Jun at Ser63/73 in Colorectal Cancer. Carcinogenesis 2011, 32, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Han, Y.; Saiz, S.; Minden, M.D. A Tumor Suppressor and Oncogene: The WT1 Story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Scharnhorst, V.; van der Eb, A.J.; Jochemsen, A.G. WT1 Proteins: Functions in Growth and Differentiation. Gene 2001, 273, 141–161. [Google Scholar] [CrossRef]
- Simpson, L.A.; Burwell, E.A.; Thompson, K.A.; Shahnaz, S.; Chen, A.R.; Loeb, D.M. The Antiapoptotic Gene A1/BFL1 Is a WT1 Target Gene That Mediates Granulocytic Differentiation and Resistance to Chemotherapy. Blood 2006, 107, 4695–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Li, X.; Zhang, D.; Xu, B.; Hu, W.; Zheng, X.; Zhu, D.; Zhou, Q.; Jiang, J.; Wu, C.; et al. IL-1β-Mediated Up-Regulation of WT1D via MiR-144-3p and Their Synergistic Effect with NF-ΚB/COX-2/HIF-1α Pathway on Cell Proliferation in LUAD. Cell. Physiol. Biochem. 2018, 48, 2493–2502. [Google Scholar] [CrossRef]
- Fridman, E.; Pinthus, J.H.; Kopolovic, J.; Ramon, J.; Mor, O.; Mor, Y. Expression of Cyclooxygenase-2 in Wilms Tumor: Immunohistochemical Study Using Tissue Microarray Methodology. J. Urol. 2006, 176, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- Maturu, P.; Jones, D.; Ruteshouser, E.C.; Hu, Q.; Reynolds, J.M.; Hicks, J.; Putluri, N.; Ekmekcioglu, S.; Grimm, E.A.; Dong, C.; et al. Role of Cyclooxygenase-2 Pathway in Creating an Immunosuppressive Microenvironment and in Initiation and Progression of Wilms’ Tumor. Neoplasia 2017, 19, 237–249. [Google Scholar] [CrossRef] [PubMed]
- George, D.P. P53 how crucial is its role in cancer? Int. J. Curr. Pharm. Res. 2011, 3, 19–25. [Google Scholar]
- Tomicic, M.T.; Dawood, M.; Efferth, T. Epigenetic alterations upstream and downstream of p53 signaling in colorectal carcinoma. Cancers 2021, 13, 4072. [Google Scholar] [CrossRef]
- Leber, M.F.; Efferth, T. Molecular Principles of Cancer Invasion and Metastasis (Review). Int. J. Oncol. 2009, 34, 881–895. [Google Scholar]
- Han, J.A.; Kim, J.I.; Ongusaha, P.P.; Hwang, D.H.; Ballou, L.R.; Mahale, A.; Aaronson, S.A.; Lee, S.W. p53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 2002, 21, 5635–5644. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.M.; Heo, J.I.; Oh, J.Y.; Kim, Y.M.; Ha, K.S.; Kim, J.I.; Han, J.A. COX-2 regulates p53 activity and inhibits DNA damage-induced apoptosis. Biochem. Biophys. Res. Commun. 2005, 328, 1107–1112. [Google Scholar] [CrossRef]
- de Moraes, E.; Dar, N.A.; Gallo, C.V.D.M.; Hainaut, P. Cross-talks between cyclooxygenase-2 and tumor suppressor protein p53: Balancing life and death during inflammatory stress and carcinogenesis. Int. J. Cancer 2007, 121, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, A.Y.; Ye, X.; Li, B.; Rojanasakul, Y.; Rankin, G.O.; Chen, Y.C. Myricetin inhibits proliferation of cisplatin-resistant cancer cells through a p53-dependent apoptotic pathway. Int. J. Oncol. 2015, 47, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhough, A.; Wallam, C.A.; Hicks, D.J.; Moorghen, M.; Williams, A.C.; Paraskeva, C. The proapoptotic BH3-only protein Bim is downregulated in a subset of colorectal cancers and is repressed by antiapoptotic COX-2/PGE2 signaling in colorectal adenoma cells. Oncogene 2010, 29, 3398–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; de Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; de Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Healy, F.M.; Prior, I.A.; MacEwan, D.J.; David MacEwan, C.J. The importance of Ras in drug resistance in cancer. Br. J. Pharmacol. 2022, 179, 2334–2355. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Weickhardt, A.J.; Tebbutt, N.C.; Mariadason, J.M. Strategies for overcoming inherent and acquired resistance to EGFR inhibitors by targeting downstream effectors in the RAS/PI3K pathway. Curr. Cancer Drug Targets 2010, 10, 824–833. [Google Scholar] [CrossRef]
- Lee, K.W.; Kang, N.J.; Rogozin, E.A.; Kim, H.G.; Cho, Y.Y.; Bode, A.M.; Lee, H.J.; Surh, Y.J.; Bowden, G.T.; Dong, Z. Myricetin is a novel natural inhibitor of neoplastic cell transformation and MEK1. Carcinogenesis 2007, 28, 1918–1927. [Google Scholar] [CrossRef]
- Jung, S.K.; Lee, K.W.; Kim, H.Y.; Oh, M.H.; Byun, S.; Lim, S.H.; Heo, Y.S.; Kang, N.J.; Bode, A.M.; Dong, Z.; et al. Myricetin suppresses UVB-induced wrinkle formation and MMP-9 expression by inhibiting Raf. Biochem. Pharmacol. 2010, 79, 1455–1461. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.K.; Lee, K.W.; Byun, S.; Lee, E.J.; Kim, J.E.; Bode, A.M.; Dong, Z.; Lee, H.J. Myricetin inhibits UVB-induced angiogenesis by regulating PI-3 kinase in vivo. Carcinogenesis 2010, 31, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas-Martínez, M.; Gutiérrez-Venegas, G. Myricetin inhibition of peptidoglycan-induced COX-2 expression in H9c2 cardiomyocytes. Prev. Nutr. Food Sci. 2019, 24, 202. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, G.; Yin, J.; Li, L.; Tan, Y.; Wei, H.; Liu, B.; Deng, L.; Tang, J.; Chen, Y.; et al. GSTP1 and cancer: Expression, methylation, polymorphisms and signaling (review). Int. J. Oncol. 2020, 56, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Nobuoka, A.; Takayama, T.; Miyanishi, K.; Sato, T.; Takanashi, K.; Hayashi, T.; Kukitsu, T.; Sato, Y.; Takahashi, M.; Okamoto, T.; et al. Glutathione-S-transferase P1-1 protects aberrant crypt foci from apoptosis induced by deoxycholic acid. Gastroenterology 2004, 127, 428–443. [Google Scholar] [CrossRef]
- Van Zanden, J.J.; Geraets, L.; Wortelboer, H.M.; Van Bladeren, P.J.; Rietjens, I.M.C.M.; Cnubben, N.H.P. Structural requirements for the flavonoid-mediated modulation of glutathione S-transferase P1-1 and GS-X pump activity in MCF7 breast cancer cells. Biochem. Pharmacol. 2004, 67, 1607–1617. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Yücer, R.; Dawood, M.; Hegazy, M.E.F.; Drif, A.; Ooko, E.; Kadioglu, O.; Seo, E.J.; Kamounah, F.S.; Titinchi, S.J.; et al. In silico and in vitro screening of 50 curcumin compounds as EGFR and NF-ΚB inhibitors. Int. J. Mol. Sci. 2022, 23, 3966. [Google Scholar] [CrossRef]
- Romano, A.; Khalid, S.A.; Dawood, M.; Boulos, J.C.; Wasfi, M.; Drif, A.; Bahramimehr, F.; Shahhamzehei, N.; Shan, L.; Efferth, T. Molecules identification of gedunin from a phytochemical depository as a novel multidrug resistance-bypassing tubulin inhibitor of cancer cells. Molecules 2022, 27, 5858. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Drif, A.I.; Efferth, T. Biomarker Profiling Revealed Carcinoembryonic Antigen as a Target of Artesunate in a Ductal Breast Cancer Patient. Anticancer Res. 2022, 42, 3483–3494. [Google Scholar] [CrossRef]
- Yue, G.G.L.; Gomes, A.J.; Saeed, M.E.M.; Tsui, K.Y.; Dawood, M.; Drif, A.I.; Wong, E.C.W.; Lee, W.F.; Liu, W.; Chiu, P.W.Y.; et al. Identification of Active Components in Andrographis Paniculata Targeting on CD81 in Esophageal Cancer in Vitro and in Vivo. Phytomedicine 2022, 102, 154183. [Google Scholar] [CrossRef]
- Ong, K.C.; Khoo, H.-E. Biological Effects of Myricetin; Elsevier Science Inc.: Amsterdam, The Netherlands, 1997; Volume 29. [Google Scholar]
- Xie, Y.; Wang, Y.; Xiang, W.; Wang, Q.; Cao, Y. Mini-Reviews in Medicinal Chemistry Send Orders for Reprints to Reprints@benthamscience. Net Mini-Rev. Med. Chem. 2020, 20, 123–133. [Google Scholar] [CrossRef]
- Devi, K.P.; Rajavel, T.; Habtemariam, S.; Fazel Nabavi, S.; Nabavi, S.M. Molecular Mechanisms Underlying Anticancer Effects of Myricetin. Life Sci. 2015, 142, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Tan, L.; Wang, M.; Ren, C.; Guo, C.; Yang, B.; Ren, Y.; Cao, Z.; Li, Y.; Pei, J. Myricetin: A Review of the Most Recent Research. Biomed. Pharmacother. 2021, 134, 111017. [Google Scholar] [CrossRef]
- Van Zanden, J.J.; De Mul, A.; Wortelboer, H.M.; Usta, M.; Van Bladeren, P.J.; Rietjens, I.M.C.M.; Cnubben, N.H.P. Reversal of in Vitro Cellular MRP1 and MRP2 Mediated Vincristine Resistance by the Flavonoid Myricetin. Biochem. Pharmacol. 2005, 69, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M.; Willmore, E.; Austin, C.A. The Dietary Flavonoids Myricetin and Fisetin Act as Dual Inhibitors of DNA Topoisomerases I and II in Cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2010, 696, 41–47. [Google Scholar] [CrossRef]
- Kondo, S.; Carr, B.I.; Takagi, K.; Huang, T.H.; Chou, Y.-M.; Yokoyama, K.; Itakura, K. Expression of Rat Microsomal Epoxide Hydrolase Gene during Liver Chemical Carcinogenesis. Cancer Res. 1990, 50, 6222–6228. [Google Scholar]
- Carr, B.I.; Laishes, B.A. Carcinogen-Induced Drug Resistance in Rat Hepatocytes. Cancer Res. 1981, 41, 1715–1719. [Google Scholar]
- Carr, B.I. Pleiotropic Drug Resistance in Hepatocytes Induced by Carcinogens Administered to Rats. Cancer Res. 1987, 47, 5577–5583. [Google Scholar]
- Solt, D.B.; Shklar, G. Rapid Induction of γ-Glutamyl Transpeptidase-Rich Intraepithelial Clones in 7,12-Dimethylbenz(a)Anthracene-Treated Hamster Buccal Pouch. Cancer Res. 1982, 42, 285–291. [Google Scholar]
- Nakai, Y.; Nonomura, N. Inflammation and Prostate Carcinogenesis. Int. J. Urol. 2013, 20, 150–160. [Google Scholar] [CrossRef]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical Basis of Inflammation-Induced Carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M.; Kawanishi, S. DNA Damage in Inflammation-Related Carcinogenesis and Cancer Stem Cells. Oxid. Med. Cell. Longev. 2013, 2013, 387014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feagins, L.A.; Souza, R.F.; Spechler, S.J. Carcinogenesis in IBD: Potential Targets for the Prevention of Colorectal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 297–305. [Google Scholar] [CrossRef]
- Malongane, F.; McGaw, L.J.; Mudau, F.N. The Synergistic Potential of Various Teas, Herbs and Therapeutic Drugs in Health Improvement: A Review. J. Sci. Food Agric. 2017, 97, 4679–4689. [Google Scholar] [CrossRef]
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren Classification and Individualized Chemotherapy in Gastric Cancer (Review). Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T.; Volm, M. Pharmacogenetics for Individualized Cancer Chemotherapy. Pharmacol. Ther. 2005, 107, 155–176. [Google Scholar] [CrossRef]
- Zhou, M.; Varol, A.; Efferth, T. Multi-Omics Approaches to Improve Malaria Therapy. Pharmacol. Res. 2021, 167, 105570. [Google Scholar] [CrossRef]
- Efferth, T.; Saeed, M.E.M.; Mirghani, E.; Alim, A.; Yassin, Z.; Saeed, E.; Khalid, H.E.; Daak, S. Integration of Phytochemicals and Phytotherapy into Cancer Precision Medicine. Oncotarget 2017, 8, 50284. [Google Scholar] [CrossRef] [Green Version]
- Poornima, P.; Kumar, J.D.; Zhao, Q.; Blunder, M.; Efferth, T. Network Pharmacology of Cancer: From Understanding of Complex Interactomes to the Design of Multi-Target Specific Therapeutics from Nature. Pharmacol. Res. 2016, 111, 290–302. [Google Scholar] [CrossRef]
- Kadioglu, O.; Saeed, M.; Mahmoud, N.; Azawi, S.; Mrasek, K.; Liehr, T.; Efferth, T. Identification of Potential Novel Drug Resistance Mechanisms by Genomic and Transcriptomic Profiling of Colon Cancer Cells with P53 Deletion. Arch. Toxicol. 2021, 95, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Kuete, V.; Kadioglu, O.; Börtzler, J.; Khalid, H.; Greten, H.J.; Efferth, T. Cytotoxicity of the Bisphenolic Honokiol from Magnolia Officinalis against Multiple Drug-Resistant Tumor Cells as Determined by Pharmacogenomics and Molecular Docking. Phytomedicine 2014, 21, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, G.; Dawood, M.; Klauck, S.M.; Sugimoto, Y.; Klinger, A.; Fleischer, E.; Shan, L.; Efferth, T. A Novel Moniliformin Derivative as Pan-Inhibitor of Histone Deacetylases Triggering Apoptosis of Leukemia Cells. Biochem. Pharmacol. 2021, 194, 114677. [Google Scholar] [CrossRef] [PubMed]
- Dawood, M.; Fleischer, E.; Klinger, A.; Bringmann, G.; Shan, L.; Efferth, T. Inhibition of Cell Migration and Induction of Apoptosis by a Novel Class II Histone Deacetylase Inhibitor, MCC2344. Pharmacol. Res. 2020, 160, 105076. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Saeed, M.E.M.; Mahmoud, N.; Hussein Azawi, S.S.; Rincic, M.; Liehr, T.; Efferth, T. Identification of Metastasis-Related Genes by Genomic and Transcriptomic Studies in Murine Melanoma. Life Sci. 2021, 267, 118922. [Google Scholar] [CrossRef]
- Dawood, M.; Ooko, E.; Efferth, T. Collateral Sensitivity of Parthenolide via NF-ΚB and HIF-α Inhibition and Epigenetic Changes in Drug-Resistant Cancer Cell Lines. Front. Pharmacol. 2019, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, M.; Manadas, B.; Efferth, T.; Cabral, C. Chemoprevention and Therapeutic Role of Essential Oils and Phenolic Compounds: Modeling Tumor Microenvironment in Glioblastoma. Pharmacol. Res. 2021, 169, 105638. [Google Scholar] [CrossRef]
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and Related Natural Flavones Are Inhibitors of CDK9 That Induce Apoptosis in Cancer Cells by Transcriptional Suppression of Mcl-1. Cell Death Dis. 2011, 2, e182. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhao, J.; Zu, Y.; Fu, Y.; Liang, L.; Luo, M.; Wang, W.; Efferth, T. Antioxidant Properties, Superoxide Dismutase and Glutathione Reductase Activities in HepG2 Cells with a Fungal Endophyte Producing Apigenin from Pigeon Pea [Cajanus cajan (L.) Millsp.]. Food Res. Int. 2012, 49, 147–152. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compounds | LBE (kcal/mol) | pKi (µM) | Pharmacophore |
|---|---|---|---|---|
| 1. | β-Sitosterol | −11.73 ± 0.33 | 0.003 ± 0.002 | ALA199, ALA202, GLN203, THR206, HIS207, PHE210, THR212, HIS214, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 2 | Celecoxib | −10.18 ± 0.03 | 0.03 ± <0.01 | HIS90, GLN192, LEU352, SER353, TYR355, LEU359, TRP387, VAL523, ALA527 |
| 3 | β-Amirin | −9.59 ± 0.01 | 0.09 ± <0.01 | LEU145, GLY225, HIS226, GLY227, VAL228, ASN375, ARG376, GLY533, ASN537, VAL538 |
| 4 | (+)-Catechin | −9.43 ± <0.01 | 0.12 ± <0.01 | ALA199, ALA202, GLN203, THR206, HIS207, PHE210, ASN382, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 5 | α-Bisabolol | −9.30 ± 0.03 | 0.15 ± 0.01 | ALA199, THE206, HIS207, PHE210, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 6 | Daucosterol | −9.26 ± 0.01 | 0.17 ± <0.01 | LEU145, LEU224, GLY225, HIS226, GLY227, VAL228, ASP229, GLY235, GLU236, THR237, LU238, ARG333, GLN374, ASN375, ARG376, ASN537, VAL538 |
| 7 | β-Eudesmol | −9.18 ± 0.01 | 0.19 ± <0.01 | ALA199, ALA202, GLN203, THR206, HIS207, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 8 | Bisabelol oxide B | −9.02 ± 0.01 | 0.24 ± <9.91 | ALA199, ALA202, GLN203, THR206, PHE210, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 9 | Kaempferol | −8.93 ± 0.02 | 0.28 ± 0.01 | ALA199, ALA202, GLN203, THR206, HIS205, ASN382, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 10 | Luteolin-7-O-glucoside | −8.92 ± 0.07 | 0.29 ± 0.03 | PHE200, GLN203, HIS207, PHE210, ASN382, TYR385, TRP387, HIS388, LEU390, LEU391, TYR404, VAL444 |
| 11 | (-)-Epicatechin | −8.89 ± 0.07 | 0.31 ± 0.04 | ALA199, ALA202, GLN203, HIS207, PHE210, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 12 | Apigenin | −8.84 ± 0.06 | 0.33 ± 0.03 | ALA199, ALA202, GLN203, HIS207, PHE210, THR212, HIS214, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390 |
| 13 | Quercitin hydrate | −8.82 ± 0.08 | 0.34 ± 0.04 | ALA199, ALA202, GLN203, THR206, HIS207, PHE210, ASN382, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 14 | Chlorogenic acid | −8.67 ± 0.10 | 0.44 ± 0.08 | ALA199, ALA202, GLN203, THR206, PHE210, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 15 | Luteolin | −8.67 ± 0.12 | 0.45 ± 0.09 | ALA202, GLN203, HIS207, PHE210, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 16 | Lupeol | −8.47 ± <0.01 | 0.62 ± <0.01 | GLY225, HIS226, GLY227, VAL228, ASP229, ARG333, ILE337, TYR373, GLN374, ASN375, GLY536, ASN537, VAL538 |
| 17 | Bisabolol oxide A | −8.38 ± <0.01 | 0.71 ± 0.01 | ALA199, PHE200, ALA202, GLN203, THR206, HIS207, TYR385, TRP387, LEU390, LEU391 |
| 18 | Guaiazulene | −8.33 ± <0.01 | 0.79 ± <0.01 | ALA199, ALA202, GLN203, HIS207, PHE210, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 19 | Myrecitin | −8.32 ± 0.41 | 0.94 ± 0.71 | GLY225, GLY227, VAL228, GLN374, ASN375, ARG376, GLY533, ASN537 |
| 20 | Quercitrin | −8.29 ± 0.06 | 0.85 ± 0.08 | ILE124, ASP125, PRO128, THR129, THR149, ALA151, ASN375, ARG376, ALA378, PHE529 |
| 21 | Farnesol | −7.98 ± 0.10 | 1.42 ± 0.23 | ALA202, THR206, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 22 | Bisabolone oxide A | −7.92 ± <0.01 | 1.57 ± <0.01 | ALA199, ALA202, GLN203, THR206, PHE210, TYR385, TRP387, HIS388, LEU390, LEU391 |
| 23 | Chamazulene | −7.74 ± 0.01 | 2.10 ± 0.01 | ALA199, ALA202, GLN203, THR206, HIS207, PHE210, ASN382, TYR385, HIS386, TRP387, HIS388, LEU390, LEU391 |
| 24 | Caffeic acid | −7.09 ± 0.08 | 6.41 ± 0.87 | ALA202, THR206, TYR385, HIS386, TRP387, HIS388, LEU391 |
| 25 | (+)-Terpinen-4-ol | −6.92 ± <0.01 | 8.47 ± 0.01 | ALA202, GLN203, HIS207, PHE210, THR212, ASN382, TYR385, HIS386, TRP387, LEU390 |
| 26 | Citronellol | −6.01 ± 0.01 | 39.50 ± 6.52 | ILE124, ASP125, THR129, THR149, ARG150, ASN375, ARG376, ILE377, ALA378, PHE529 |
| 27 | P-Cymene | −6.01 ± <0.01 | 39.57 ± <0.01 | ALA202, THR206, TYR385, HIS388, LEU390, LEU391, ALA199, GLN203, THR206, HIS207, PHE210, TYR385, TRP387, HIS388, LEU390, LYS97, ASN104, GLN350, TYR355, HIS356, LYS358 |
| Myricetin (log10 IC50, M) | Control Drug (log10 IC50, M) | ||
|---|---|---|---|
| ABCB1 Expression | Epirubicin | ||
| 7q21 (Chromosomal | r-value | −0.120 | 0.447 * |
| Locus of ABCB1 Gene) | p-value | 0.207 | 3.55 × 10−4 * |
| ABCB1 Expression | r-value | −0.124 | 0.533 * |
| (Microarray) | p-value | 0.186 | * 6.82 × 10−6 |
| ABCB1 Expression | r-value | 0.118 | * 0.410 |
| (RT-PCR) | p-value | 0.215 | * 1.54 × 10−3 |
| ABCB5 Expression | Maytansine | ||
| ABCB5 Expression | r-value | −0.040 | 0.454 * |
| (Microarray) | p-value | 0.384 | 6.67 × 10−4 * |
| ABCB5 Expression | r-value | 0.060 | 0.402 * |
| (RT-PCR) | p-value | 0.330 | 0.0026 * |
| ABCC1 Expression | Vinblastine | ||
| DNA Gene | r-value | 0.059 | 0.429 * |
| Copy Number | p-value | 0.333 | 0.001 * |
| ABCC1 Expression | r-value | −0.035 | 0.398 * |
| (Microarray) | p-value | 0.402 | 0.003 * |
| ABCC1 Expression | r-value | 0.149 | 0.299 |
| (RT-PCR) | p-value | 0.170 | 0.036 * |
| ABCG2 Expression | Pancratistatin | ||
| ABCG2 Expression | r-value | 0.163 | 0.329 * |
| (Microarray) | p-value | 0.120 | 0.006 * |
| ABCG2 Expression | r-value | −0.127 | 0.346 * |
| (Western Blot) | p-value | 0.177 | 0.004 * |
| EGFR Expression | Erlotinib | ||
| EGFR Gene | r-value | 0.135 | −0.245 |
| Copy Number | p-value | 0.160 | 0.029 * |
| EGFR Expression | r-value | 0.133 | −0.458 * |
| (Microarray) | p-value | 0.164 | 1.15 × 10−4 * |
| EGFR Expression | r-value | 0.077 | −0.379 * |
| (PCR Slot Blot) | p-value | 0.291 | 0.002 * |
| EGFR Expression | r-value | 0.166 | −0.376 * |
| (Protein Array) | p-value | 0.113 | 0.001 * |
| N-/K-/H-RAS Mutations | Melphalan | ||
| TP53 Mutation | r-value | 0.052 | 0.367 * |
| (cDNA Sequencing) | p-value | 0.354 | 0.002 * |
| TP53 Mutation | 5-Fluorouracil | ||
| TP53 Mutation | r-value | 0.077 | −0.502 * |
| (cDNA Sequencing) | p-value | 0.290 | 3.50 × 10−5 * |
| TP53 Function | r-value | 0.211 | −0.436 * |
| (Yeast Functional Assay) | p-value | 0.071 | 5.49 × 10−4 * |
| WT1 Expression | Ifosfamide | ||
| WT1 Expression | r-value | 0.064 | −0.316 * |
| (Microarray) | p-value | 0.320 | 0.007 * |
| GSTP1 Expression | Etoposide | ||
| GSTP1 Expression | r-value | −0.257 | 0.399 |
| (Microarray) | p-value | 0.028 | 9.58 × 10−4 * |
| GST Expression | r-value | −0.225 | 0.509 |
| (Northern Blot) | p-value | 0.048 | 2.24 × 10−5 * |
| HSP90 Expression | Geldanamycin | ||
| HSP90 Expression | r-value | −0.172 | −0.392 * |
| (Microarray) | p-value | 0.105 | 0.001 * |
| Proliferation | 5-Fluorouracil | ||
| Cell Doubling | r-value | 0.258 | 0.627 * |
| p-value | 0.031 | 7.14 × 10−6 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drif, A.I.; Avula, B.; Khan, I.A.; Efferth, T. COX2-Inhibitory and Cytotoxic Activities of Phytoconstituents of Matricaria chamomilla L. Appl. Sci. 2023, 13, 8935. https://doi.org/10.3390/app13158935
Drif AI, Avula B, Khan IA, Efferth T. COX2-Inhibitory and Cytotoxic Activities of Phytoconstituents of Matricaria chamomilla L. Applied Sciences. 2023; 13(15):8935. https://doi.org/10.3390/app13158935
Chicago/Turabian StyleDrif, Assia I., Bharathi Avula, Ikhlas A. Khan, and Thomas Efferth. 2023. "COX2-Inhibitory and Cytotoxic Activities of Phytoconstituents of Matricaria chamomilla L." Applied Sciences 13, no. 15: 8935. https://doi.org/10.3390/app13158935
APA StyleDrif, A. I., Avula, B., Khan, I. A., & Efferth, T. (2023). COX2-Inhibitory and Cytotoxic Activities of Phytoconstituents of Matricaria chamomilla L. Applied Sciences, 13(15), 8935. https://doi.org/10.3390/app13158935