



A Novel Deep Learning Method for Predicting RNA-Protein Binding Sites

Abstract
:1. Introduction
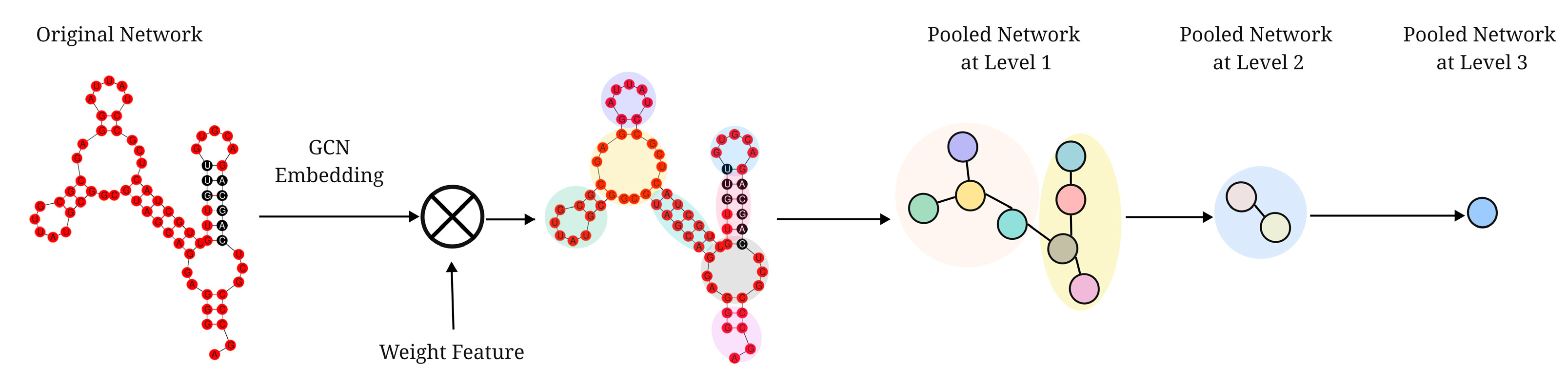
- HPNet uses DiffPool, a hierarchical pooling network, to discover hierarchical features of RNA secondary structure. It divides the substructures of the secondary structure into the same cluster and learns more meaningful graph-level embeddings;
- HPNet recognizes binding sequences and automatically extracts binding motifs using the CNN and DiffPool. It can determine whether binding sites exist and capture binding motifs without domain knowledge. The area under the curve (AUC) for HPNet was found to be significantly better than the state-of-the-art prediction method;
- A context-average debiasing method is proposed. In response to the traditional debiasing method of replacing clip sites with random nucleotides, this paper proposes a debiasing method of replacing clip sites with average-context features of clip sites.
2. Materials and Methods
2.1. Datasets and Data Processing
2.2. Sequence Coding
2.3. Secondary Structure Coding
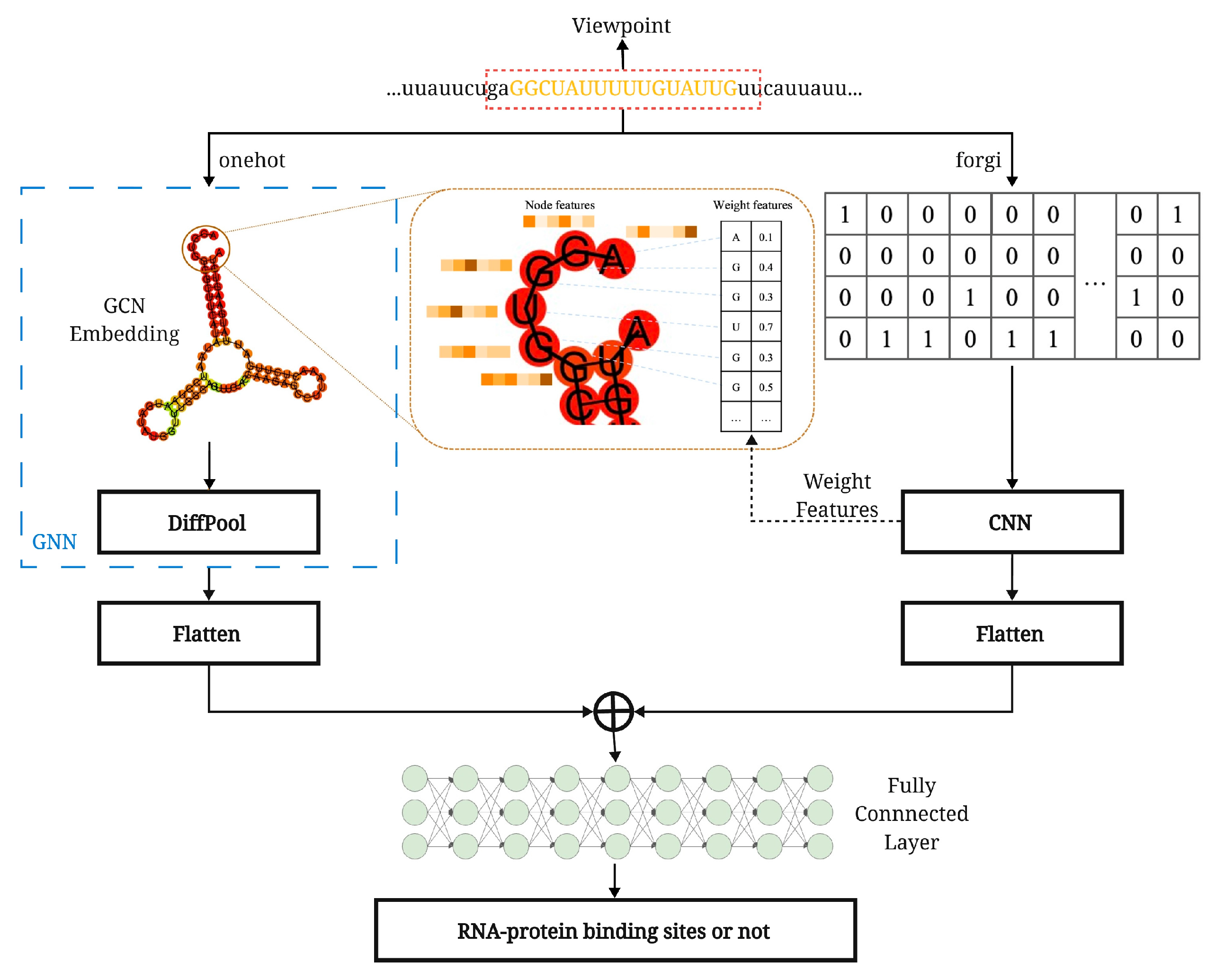
2.4. HPNet Architecture
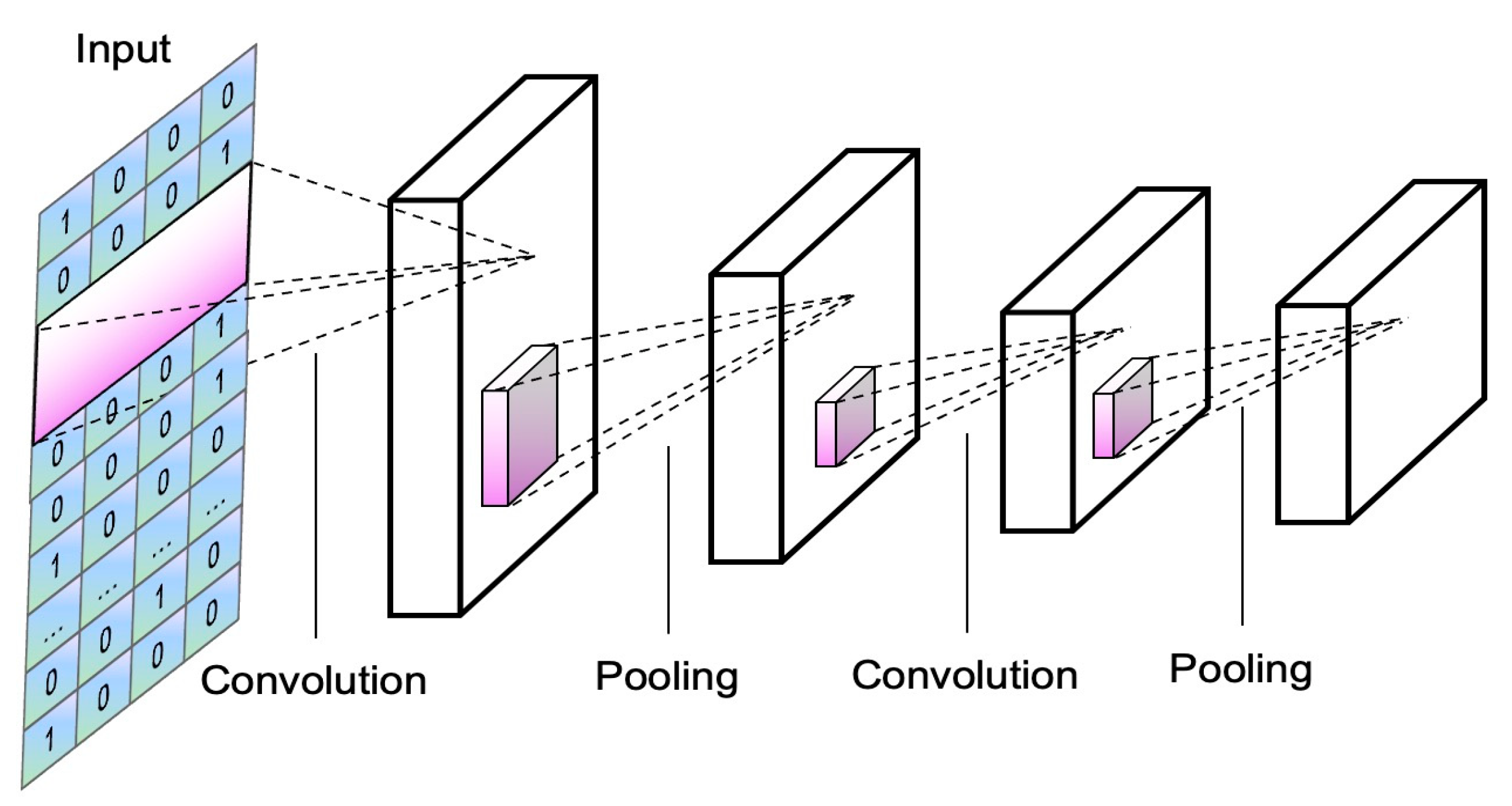
2.5. The Convolutional Neural Network in HPNet
2.6. DiffPool in HPNet
2.7. Fully Connected Layer and Loss Function
2.8. Debiasing Data
2.9. Parameter Optimization
3. Results
3.1. Baseline
- GraphProt: This method uses a hypergraph to describe the RNA secondary structure fully and uses a graph kernel approach to extract features from the hypergraph. The model is then trained through machine learning based on the features to predict RNA-binding sites;
- Deepnet-rbp [41]: This method added the RNA tertiary structure to the prediction model for the first time. It involves constructing a framework containing the RNA sequence, secondary structure, and tertiary structure, which represent the specificities of the RBP-binding sites in three dimensions, respectively;
- iDeppE: This method uses two CNNs to learn global and local RNA sequence features. For the global CNN, an entire sequence is padded to the same length and used directly as the input. For the local CNN, an RNA sequence is split into multiple overlapping fixed-length subsequences, and each sub-sequence is used as a separate signal channel of the full-length sequence. The two CNNs are trained independently and then combined for prediction;
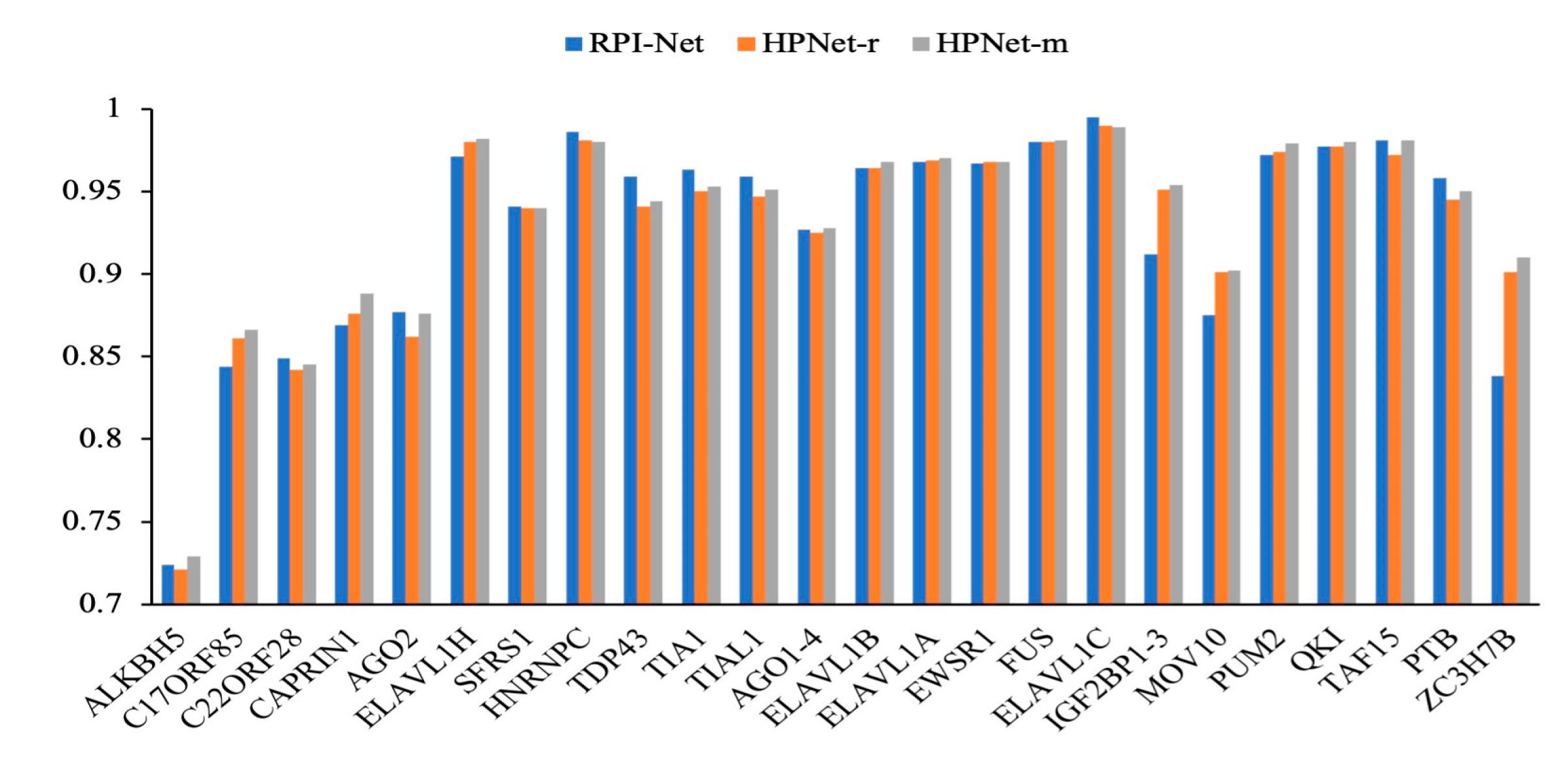
- RPI-Net: This method introduces GCN and LSTM deep-learning models. The graphical representation of RNA secondary structure can be effectively learned using the GCN and message passing takes place through the LSTM. The learned graphical features are then inputted into the Bi-LSTM to learn the global embedding. An end-to-end learning method is implemented;
- DeepPN: This method consists of a CNN and a GCN, a parallel learning network. The CNN and GCN are used to learn sequence features from different perspectives to compensate for the lack of features.
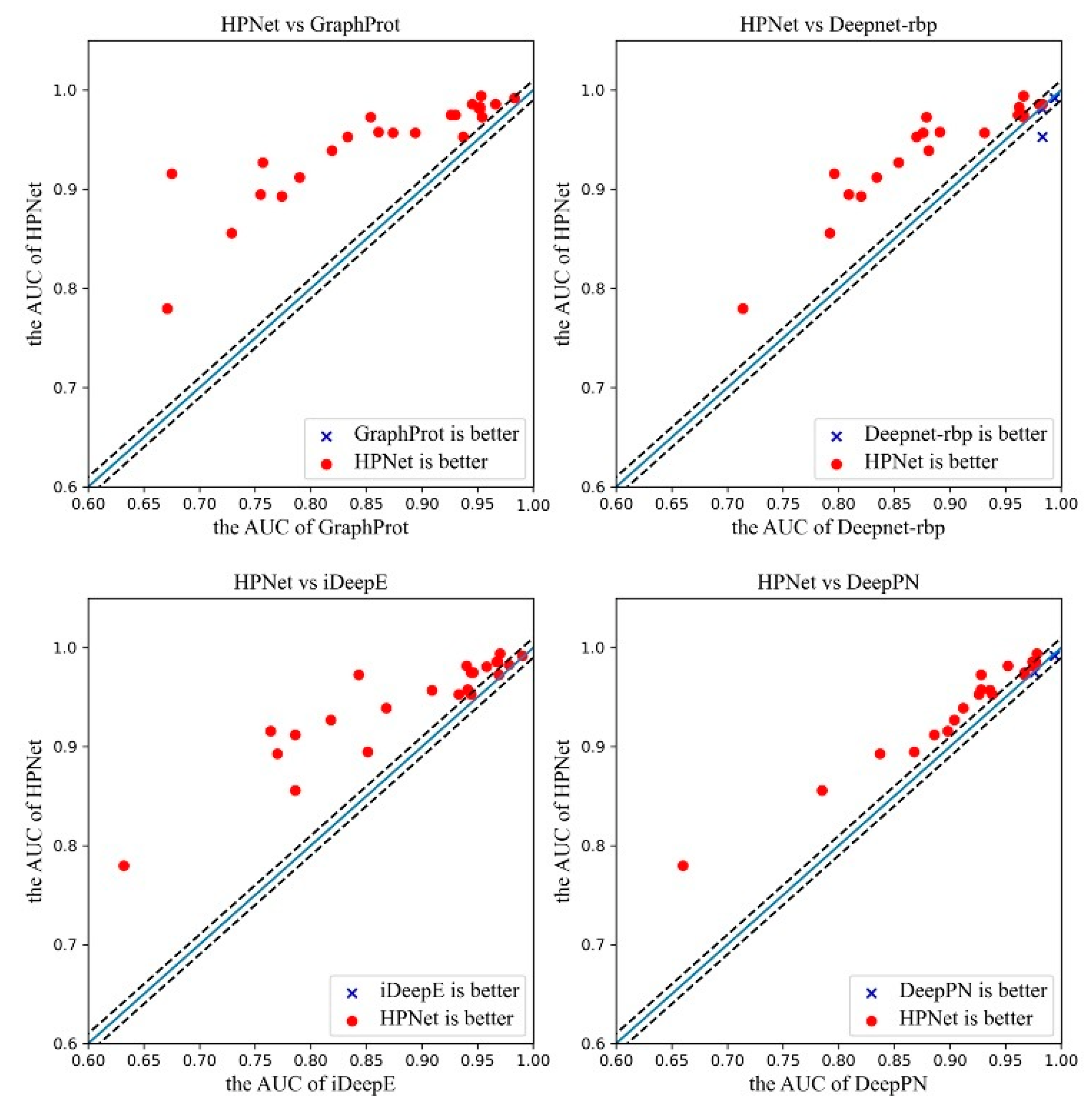
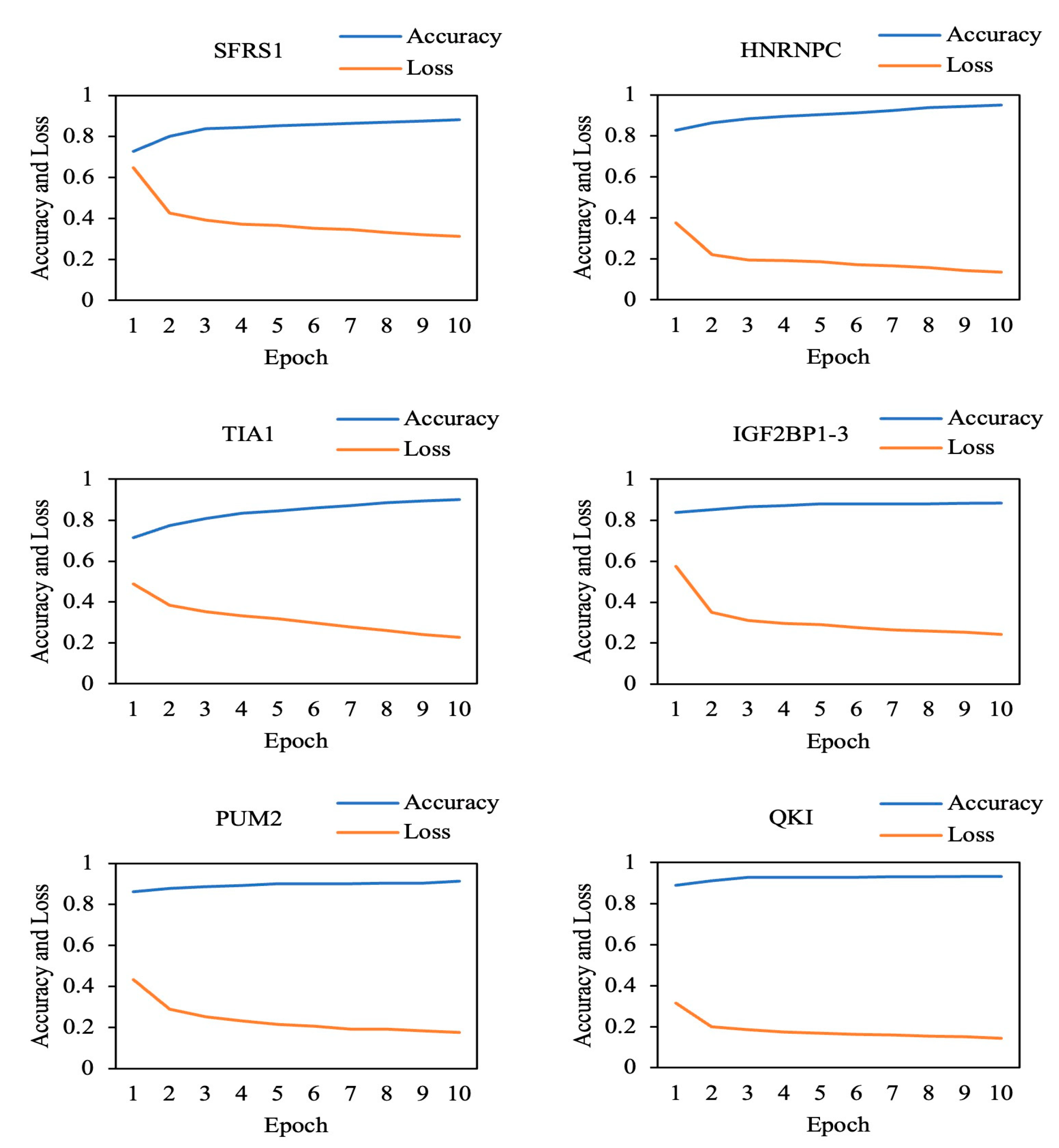
3.2. Prediction Results for the Original Dataset
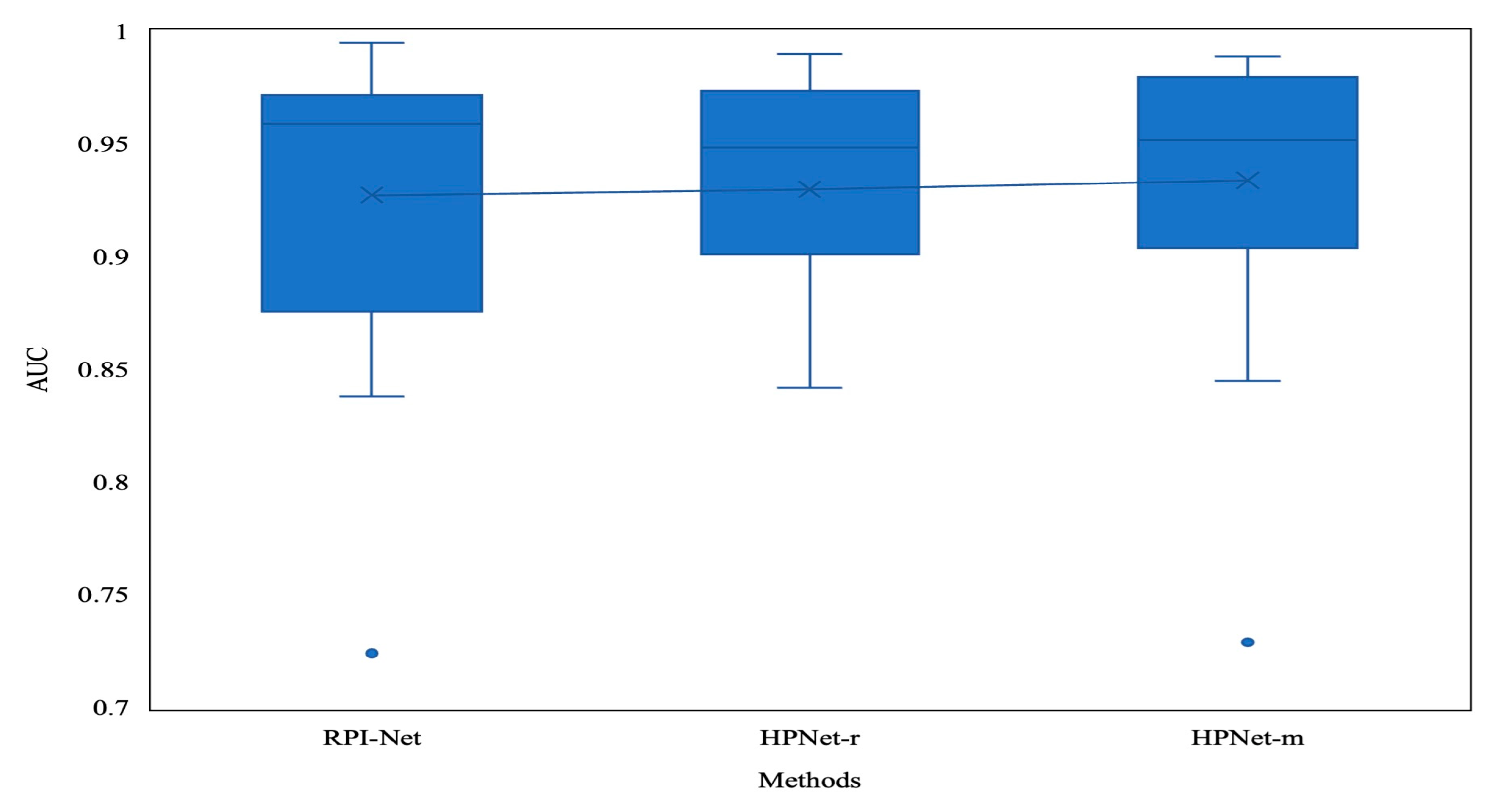
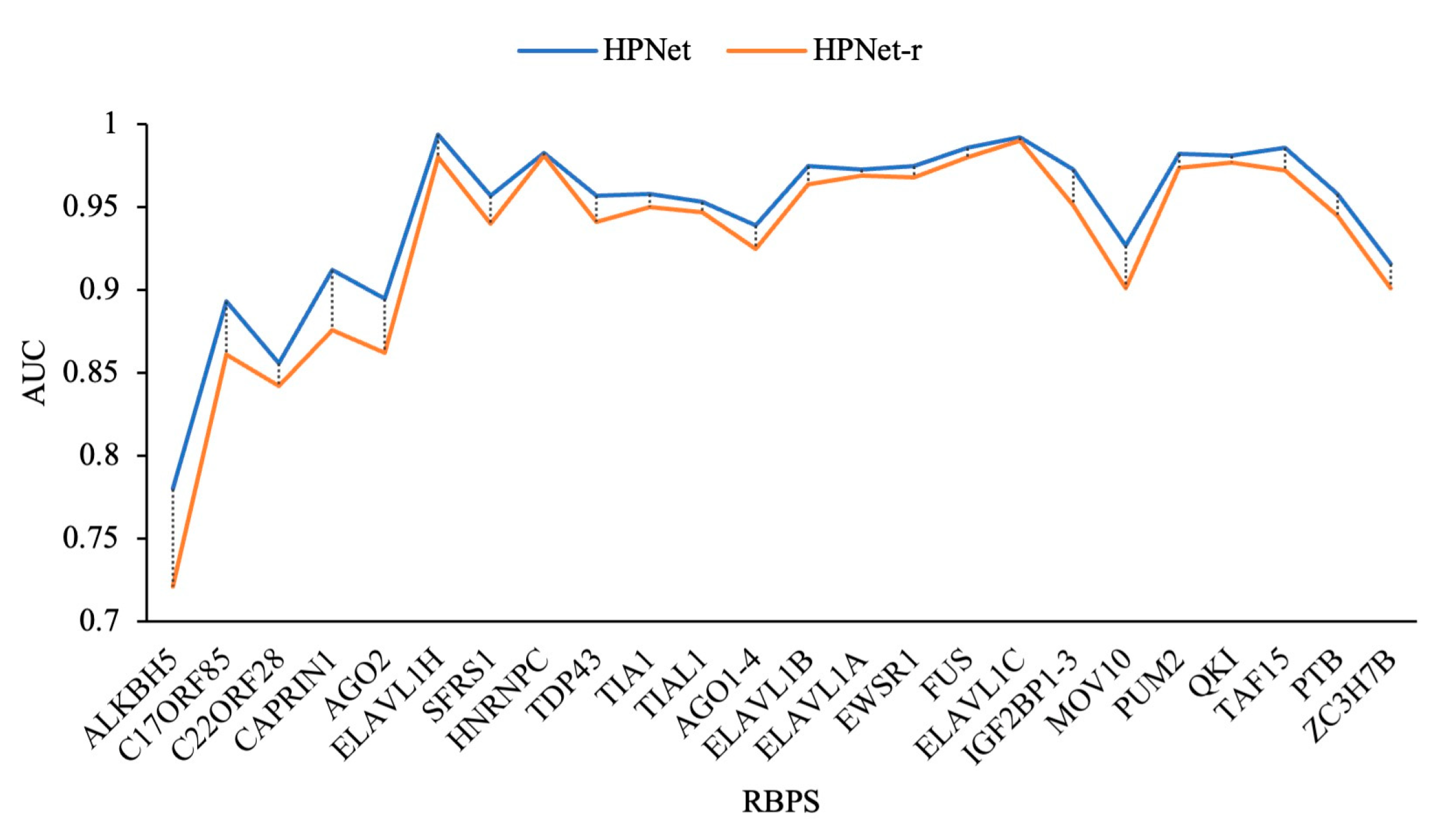
3.3. Prediction Results for the Debiased Datasets
3.4. Motif Visualizations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McHugh, C.A.; Russell, P.; Guttman, M. Methods for Comprehensive Experimental Identification of RNA-Protein Interactions. Genome Biol. 2014, 15, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breaker, R.R.; Joyce, G.F. The Expanding View of RNA and DNA Function. Chem. Biol. 2014, 21, 1059–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achsel, T.; Bagni, C. Cooperativity in RNA–Protein Interactions: The Complex Is More than the Sum of Its Partners. Curr. Opin. Neurobiol. 2016, 39, 146–151. [Google Scholar] [CrossRef]
- Oliveira, C.; Faoro, H.; Alves, L.R.; Goldenberg, S. RNA-Binding Proteins and Their Role in the Regulation of Gene Expression in Trypanosoma Cruzi and Saccharomyces Cerevisiae. Genet. Mol. Biol. 2017, 40, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Re, A.; Joshi, T.; Kulberkyte, E.; Morris, Q.; Workman, C.T. RNA-Protein Interactions: An Overview. Methods Mol. Biol. 2014, 1097, 491–521. [Google Scholar] [CrossRef]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to Study RNA–Protein Interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef]
- Cozzolino, F.; Iacobucci, I.; Monaco, V.; Monti, M. Protein–DNA/RNA Interactions: An Overview of Investigation Methods in the -Omics Era. J. Proteome Res. 2021, 20, 3018–3030. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, Y.; Wu, T.; Zhu, T.; Ji, X.; Kwon, Y.-S.; Zhang, C.; Yeo, G.; Black, D.L.; Sun, H.; et al. Genome-Wide Analysis of PTB-RNA Interactions Reveals a Strategy Used by the General Splicing Repressor to Modulate Exon Inclusion or Skipping. Mol. Cell 2009, 36, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-Binding Proteins in Human Genetic Disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef]
- Ascano, M.; Hafner, M.; Cekan, P.; Gerstberger, S.; Tuschl, T. Identification of RNA–Protein Interaction Networks Using PAR-CLIP. Wiley Interdiscip. Rev. RNA 2012, 3, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Nechay, M.; Kleiner, R.E. High-Throughput Approaches to Profile RNA-Protein Interactions. Curr. Opin. Chem. Biol. 2020, 54, 37–44. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Zarnack, K.; Luscombe, N.M.; Ule, J. Protein–RNA Interactions: New Genomic Technologies and Perspectives. Nat. Rev. Genet. 2012, 13, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-Wide Identification of Polycomb-Associated RNAs by RIP-Seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanegas, P.L.; Hudson, G.A.; Davis, A.R.; Kelly, S.C.; Kirkpatrick, C.C.; Znosko, B.M. RNA CoSSMos: Characterization of Secondary Structure Motifs—A Searchable Database of Secondary Structure Motifs in RNA Three-Dimensional Structures. Nucleic Acids Res. 2012, 40, D439–D444. [Google Scholar] [CrossRef] [Green Version]
- Balcerak, A.; Trebinska-Stryjewska, A.; Konopinski, R.; Wakula, M.; Grzybowska, E.A. RNA-Protein Interactions: Disorder, Moonlighting and Junk Contribute to Eukaryotic Complexity. Open Biol. 2019, 9, 190096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolma, A.; Zhang, J.; Mondragón, E.; Morgunova, E.; Kivioja, T.; Laverty, K.U.; Yin, Y.; Zhu, F.; Bourenkov, G.; Morris, Q.; et al. Binding Specificities of Human RNA-Binding Proteins toward Structured and Linear RNA Sequences. Genome Res. 2020, 30, 962–973. [Google Scholar] [CrossRef]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The Graph Neural Network Model. IEEE Trans. Neural Netw. 2009, 20, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Dai, Q.; Song, J.; Duan, X.; Yang, H.; Yang, Z. Predicting RBP Binding Sites of RNA With High-Order Encoding Features and CNN-BLSTM Hybrid Model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2409–2419. [Google Scholar] [CrossRef]
- Macke, T.J.; Ecker, D.J.; Gutell, R.R.; Gautheret, D.; Case, D.A.; Sampath, R. RNAMotif, an RNA Secondary Structure Definition and Search Algorithm. Nucleic Acids Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Chełkowska-Pauszek, A.; Kosiński, J.G.; Marciniak, K.; Wysocka, M.; Bąkowska-Żywicka, K.; Żywicki, M. The Role of RNA Secondary Structure in Regulation of Gene Expression in Bacteria. Int. J. Mol. Sci. 2021, 22, 7845. [Google Scholar] [CrossRef] [PubMed]
- Kazan, H.; Ray, D.; Chan, E.T.; Hughes, T.R.; Morris, Q. RNAcontext: A New Method for Learning the Sequence and Structure Binding Preferences of RNA-Binding Proteins. PLoS Comput. Biol. 2010, 6, e1000832. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, Y.; Wang, Y.; Berger, B. RCK: Accurate and Efficient Inference of Sequence- and Structure-Based Protein–RNA Binding Models from RNAcompete Data. Bioinformatics 2016, 32, i351–i359. [Google Scholar] [CrossRef] [Green Version]
- Maticzka, D.; Lange, S.J.; Costa, F.; Backofen, R. GraphProt: Modeling Binding Preferences of RNA-Binding Proteins. Genome Biol. 2014, 15, R17. [Google Scholar] [CrossRef] [Green Version]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the Sequence Specificities of DNA- and RNA-Binding Proteins by Deep Learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Shen, H.-B. Predicting RNA-Protein Binding Sites and Motifs through Combining Local and Global Deep Convolutional Neural Networks. Bioinformatics 2018, 34, 3427–3436. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Rijnbeek, P.; Yan, J.; Shen, H.-B. Prediction of RNA-Protein Sequence and Structure Binding Preferences Using Deep Convolutional and Recurrent Neural Networks. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liu, Y.; Shi, Y.; Zhang, W.; Yang, C.; Liu, H. Deep Neural Networks for Inferring Binding Sites of RNA-Binding Proteins by Using Distributed Representations of RNA Primary Sequence and Secondary Structure. BMC Genom. 2020, 21, 866. [Google Scholar] [CrossRef]
- Yan, J.; Zhu, M. A Review About RNA–Protein-Binding Sites Prediction Based on Deep Learning. IEEE Access 2020, 8, 150929–150944. [Google Scholar] [CrossRef]
- Yan, Z.; Hamilton, W.L.; Blanchette, M. Graph Neural Representational Learning of RNA Secondary Structures for Predicting RNA-Protein Interactions. Bioinformatics 2020, 36, i276–i284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tong, H.; Xu, J.; Maciejewski, R. Graph Convolutional Networks: A Comprehensive Review. Comput. Soc. Netw. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, B.; Wang, Z.; Lehnert, K.; Gahegan, M. DeepPN: A Deep Parallel Neural Network Based on Convolutional Neural Network and Graph Convolutional Network for Predicting RNA-Protein Binding Sites. BMC Bioinform. 2022, 23, 257. [Google Scholar] [CrossRef] [PubMed]
- Anders, G.; Mackowiak, S.D.; Jens, M.; Maaskola, J.; Kuntzagk, A.; Rajewsky, N.; Landthaler, M.; Dieterich, C. DoRiNA: A Database of RNA Interactions in Post-Transcriptional Regulation. Nucleic Acids Res. 2012, 40, D180–D186. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Katsantoni, M.; Köster, T.; Marks, J.; Mukherjee, J.; Staiger, D.; Ule, J.; Zavolan, M. CLIP and Complementary Methods. Nat. Rev. Methods Prim. 2021, 1, 20. [Google Scholar] [CrossRef]
- Thiel, B.C.; Beckmann, I.K.; Kerpedjiev, P.; Hofacker, I.L. 3D Based on 2D: Calculating Helix Angles and Stacking Patterns Using Forgi 2.0, an RNA Python Library Centered on Secondary Structure Elements. F1000Research 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA Base Pairing Probabilities in Large Sequences. Bioinformatics 2006, 22, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-Based Learning Applied to Document Recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Ying, R.; You, J.; Morris, C.; Ren, X.; Hamilton, W.L.; Leskovec, J. Hierarchical Graph Representation Learning with Differentiable Pooling. In Proceedings of the 32nd International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; Curran Associates Inc.: Red Hook, NY, USA, 2018; pp. 4805–4815. [Google Scholar]
- Hamilton, W.L.; Ying, R.; Leskovec, J. Inductive Representation Learning on Large Graphs. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Montreal, QC, Canada, 3–8 December 2018; Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 1025–1035. [Google Scholar]
- Zhang, S.; Zhou, J.; Hu, H.; Gong, H.; Chen, L.; Cheng, C.; Zeng, J. A Deep Learning Framework for Modeling Structural Features of RNA-Binding Protein Targets. Nucleic Acids Res. 2016, 44, e32. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A Compendium of RNA-Binding Motifs for Decoding Gene Regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, E.; Bailey, T.; Grant, C.E.; Noble, W.S.; Keich, U. Improved Similarity Scores for Comparing Motifs. Bioinformatics 2011, 27, 1603–1609. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Elkan, C. Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Kota, S.K.; Lim, Z.W.; Kota, S.B. Elavl1 Impacts Osteogenic Differentiation and MRNA Levels of Genes Involved in ECM Organization. Front. Cell Dev. Biol. 2021, 9, 606971. [Google Scholar] [CrossRef] [PubMed]
- Dember, L.M.; Kim, N.D.; Liu, K.Q.; Anderson, P. Individual RNA Recognition Motifs of TIA-1 and TIAR Have Different RNA Binding Specificities. J. Biol. Chem. 1996, 271, 2783–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, D.W.; Knigge, D.M.; Gu, A.; Bekkers, E.J.; Gavves, E.; Tomczak, J.M.; Hoogendoorn, M. Towards a General Purpose CNN for Long Range Dependencies in ND. arXiv 2022, arXiv:2206.03398. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure (Abbreviation) | Description | Graphical Representation |
|---|---|---|
| Stem (S) | Consecutive nucleotide-paired regions |  |
| Hairpin loop (H) | An unpaired loop structure formed by the nucleotide strings vacated between the two nucleotide strings forming the stem region |  |
| Interior loop (I) | The internal loop can contain unpaired nucleotides on either or both strands, flanked by stems |  |
| Multiloop (M) | The single-stranded region between two stems |  |
| Fiveprime (F) | The unpaired nucleotides at the 5′ end of a molecule/chain | / |
| Threeprime (T) | The unpaired nucleotides at the 3′ end of a molecule/chain | / |
| RBPs | GraphProt | Deepnet-rbp | iDeepE | DeepPN | HPNet |
|---|---|---|---|---|---|
| ALKBH5 | 0.68 | 0.714 | 0.758 | 0.66 | 0.78 |
| C17ORF85 | 0.800 | 0.820 | 0.830 | 0.837 | 0.893 |
| C22ORF28 | 0.751 | 0.792 | 0.837 | 0.785 | 0.856 |
| CAPRIN1 | 0.855 | 0.834 | 0.893 | 0.886 | 0.912 |
| AGO2 | 0.765 | 0.809 | 0.884 | 0.868 | 0.895 |
| ELAVL1H | 0.955 | 0.966 | 0.979 | 0.978 | 0.994 |
| SFRS1 | 0.898 | 0.931 | 0.946 | 0.936 | 0.957 |
| HNRNPC | 0.952 | 0.962 | 0.976 | 0.977 | 0.983 |
| TDP43 | 0.874 | 0.876 | 0.945 | 0.936 | 0.957 |
| TIA1 | 0.861 | 0.891 | 0.937 | 0.928 | 0.958 |
| TIAL1 | 0.833 | 0.870 | 0.934 | 0.926 | 0.953 |
| AGO1-4 | 0.895 | 0.881 | 0.915 | 0.912 | 0.939 |
| ELAVL1B | 0.935 | 0.961 | 0.971 | 0.976 | 0.975 |
| ELAVL1A | 0.959 | 0.966 | 0.964 | 0.967 | 0.973 |
| EWSR1 | 0.935 | 0.966 | 0.969 | 0.954 | 0.975 |
| FUS | 0.968 | 0.980 | 0.985 | 0.977 | 0.986 |
| ELAVL1C | 0.991 | 0.994 | 0.988 | 0.994 | 0.992 |
| IGF2BP1-3 | 0.889 | 0.879 | 0.947 | 0.928 | 0.973 |
| MOV10 | 0.863 | 0.854 | 0.916 | 0.904 | 0.927 |
| PUM2 | 0.954 | 0.971 | 0.967 | 0.952 | 0.982 |
| QKI | 0.957 | 0.983 | 0.970 | 0.975 | 0.981 |
| TAF15 | 0.970 | 0.983 | 0.976 | 0.974 | 0.986 |
| PTB | 0.937 | 0.983 | 0.944 | 0.938 | 0.958 |
| ZC3H7B | 0.820 | 0.796 | 0.907 | 0.898 | 0.916 |
| Mean | 0.887 | 0.902 | 0.931 | 0.919 | 0.945 |
| Method | Mean AUC |
|---|---|
| RPI-Net | 0.927 |
| HPNet-r | 0.930 |
| HPNet-m | 0.933 |
| RBPs | Motifs | RBPs | Motifs | RBPs | Motifs |
|---|---|---|---|---|---|
| HNRNPC | p-value: 3.22 × 10−5 | SRSF1 | p-value: 3.58 × 10−3 | TIA1 | p-value: 5.70 × 10−4 |
| PUM2 | p-value: 9.02 × 10−4 | QKI | p-value: 1.41 × 10−4 | IGF2BP123 | p-value: 9.66 × 10−5 |
| RBPs | Motifs | RBPs | Motifs | RBPs | Motifs | RBPs | Motifs |
|---|---|---|---|---|---|---|---|
| ALKBH5 |  | C17ORF85 |  | C22ORF28 |  | CAPRIN1 |  |
| AGO2 |  | TDP43 |  | TAF15 |  | TIAL1 |  |
| AGO1234 |  | ELAVL1A |  | ELAVL1B |  | ELAVL1C |  |
| ELAVL1H |  | EWSR1 |  | FUS |  | ZC3H7B |  |
| MOV10 |  | PTB |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Chang, F.; Lv, H.; Zou, G.; Zhang, B. A Novel Deep Learning Method for Predicting RNA-Protein Binding Sites. Appl. Sci. 2023, 13, 3247. https://doi.org/10.3390/app13053247
Zhao X, Chang F, Lv H, Zou G, Zhang B. A Novel Deep Learning Method for Predicting RNA-Protein Binding Sites. Applied Sciences. 2023; 13(5):3247. https://doi.org/10.3390/app13053247
Chicago/Turabian StyleZhao, Xueru, Furong Chang, Hehe Lv, Guobing Zou, and Bofeng Zhang. 2023. "A Novel Deep Learning Method for Predicting RNA-Protein Binding Sites" Applied Sciences 13, no. 5: 3247. https://doi.org/10.3390/app13053247
APA StyleZhao, X., Chang, F., Lv, H., Zou, G., & Zhang, B. (2023). A Novel Deep Learning Method for Predicting RNA-Protein Binding Sites. Applied Sciences, 13(5), 3247. https://doi.org/10.3390/app13053247