
Sum-Frequency-Generation-Based Laser Sidebands for Tunable Femtosecond Raman Spectroscopy in the Ultraviolet


Abstract
:
1. Introduction
2. Experimental Section

3. Results and Discussion
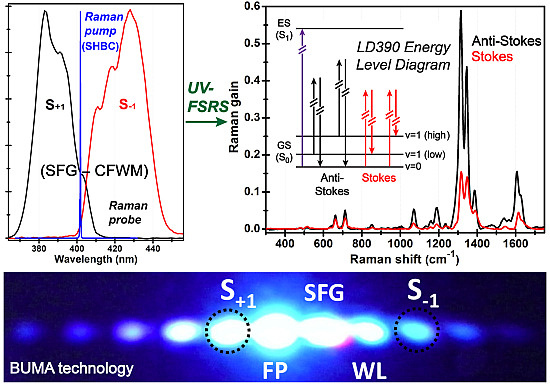
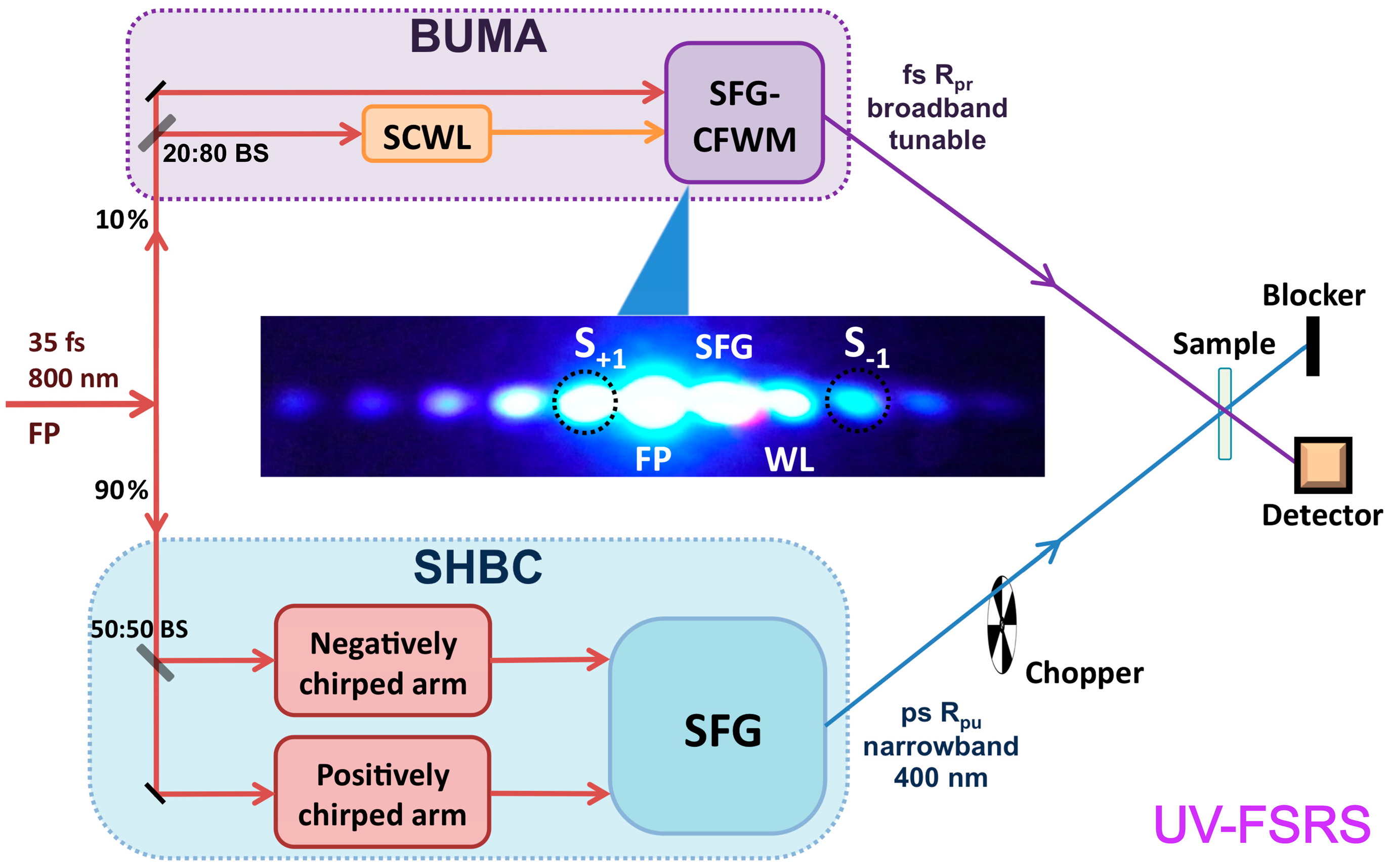
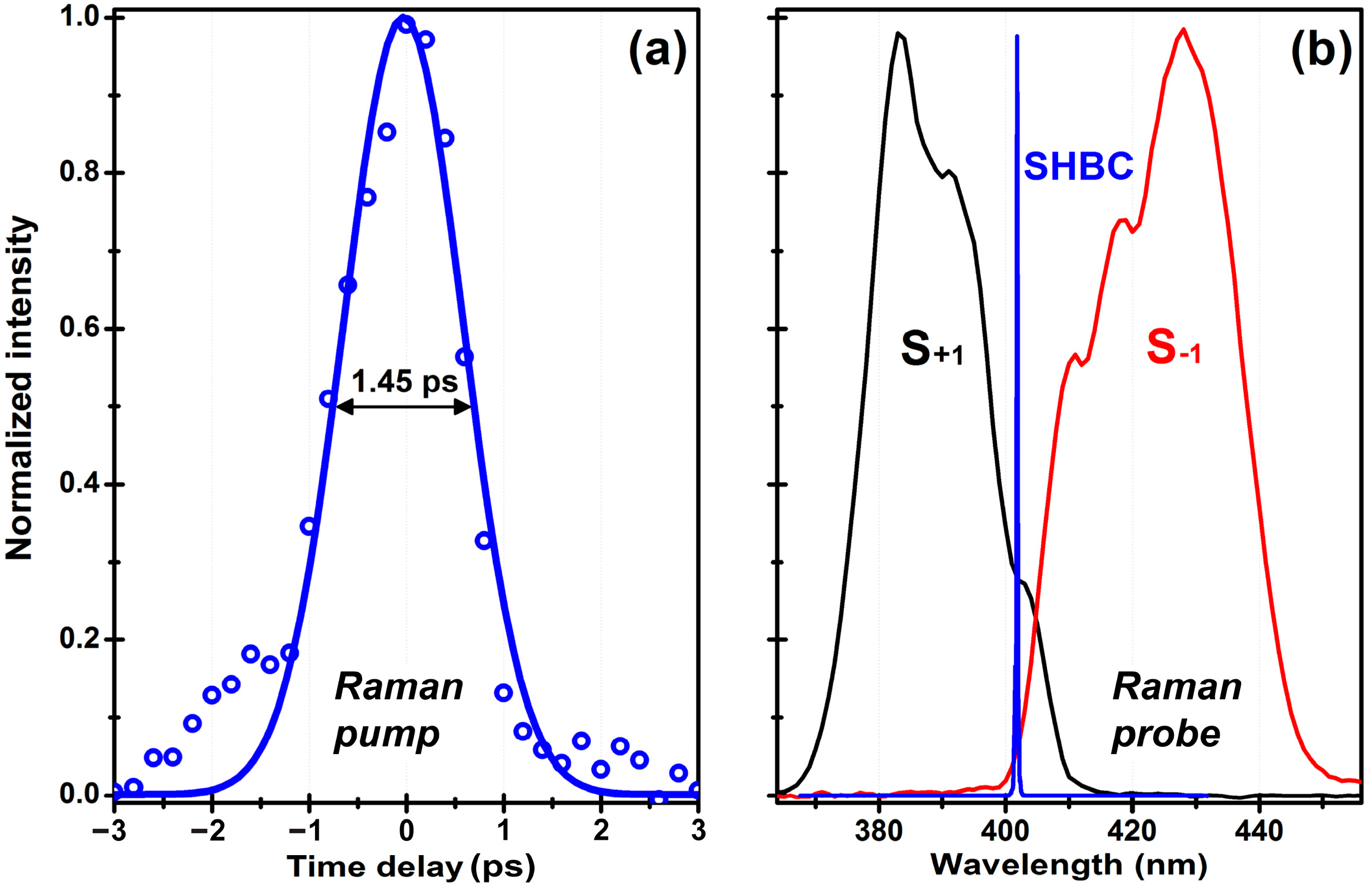
3.1. UV-FSRS Setup with SHBC and SFG-CFWM

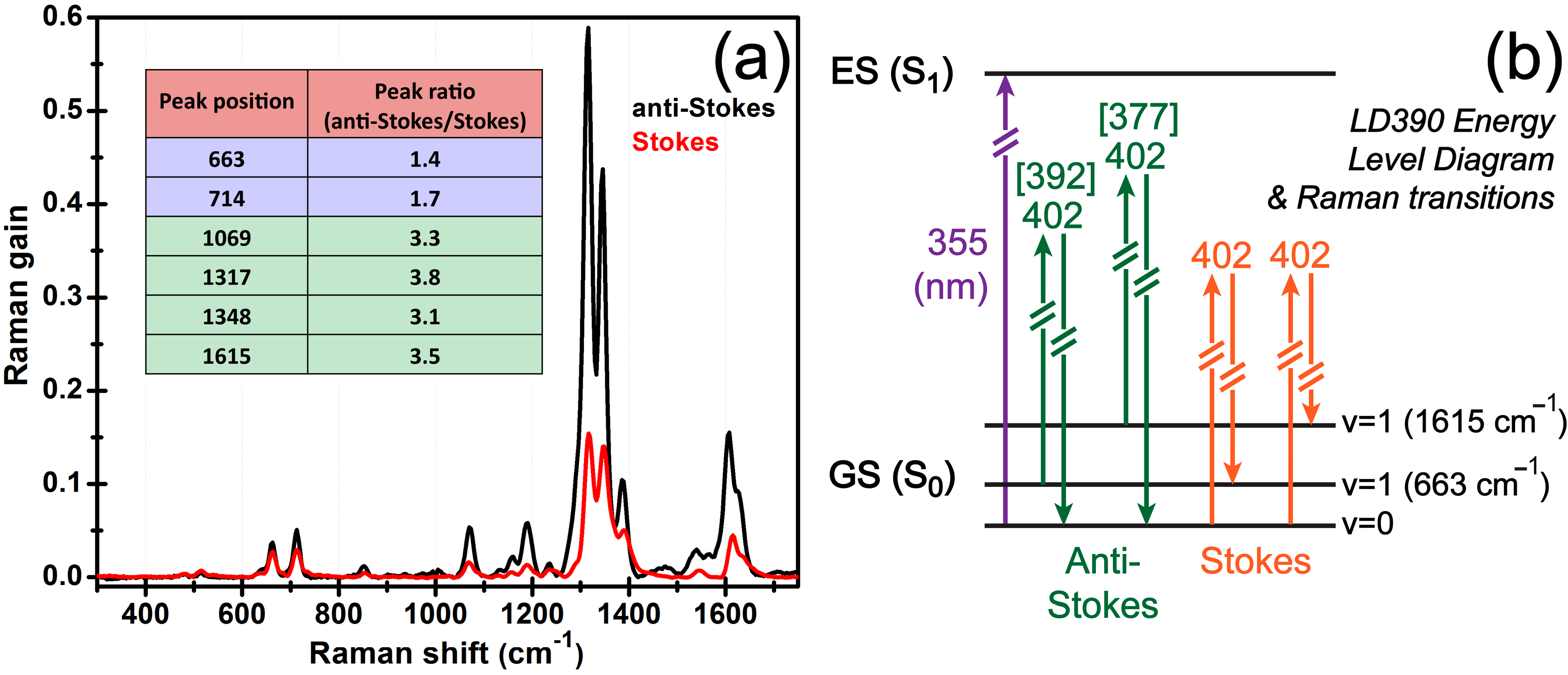
3.2. Ground-State FSRS of Laser Dye LD390

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S0 calc. a (cm–1) | S0 FSRS b (cm–1) | Vibrational mode assignment c |
|---|---|---|
| 665 | 663 | Ring in-plane asymmetric deformation |
| 711 | 714 | Ring asymmetric breathing with N1–CH3 stretching |
| 1068 | 1069 | A-ring deformation and H rocking, B-ring small-scale breathing, and N13–(CH3)2 H twisting |
| 1312 | 1317 | N1–C2 stretching with ring asymmetric deformation and ring-H rocking, and (N1)–CH3 methyl group bending |
| 1357 | 1348 | C7–N8 stretching and A-ring H rocking, A-ring in-plane deformation with some C9–C10 stretching |
| 1393 | 1390 | N1–CH3 stretching and methyl group symmetric bending, A-ring in-plane deformation, and ring-H rocking |
| 1613 | 1615 | Ring C=C and C=N stretching, ring-H rocking with C2=O11 stretching |

3.3. Comparison between the Stokes and Anti-Stokes FSRS

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zewail, A.H. Femtochemistry: Ultrafast Dynamics of the Chemical Bond; World Scientific: Singapore, 1994. [Google Scholar]
- Hochstrasser, R.M. Two-dimensional spectroscopy at infrared and optical frequencies. Proc. Natl. Acad. Sci. USA 2007, 104, 14190–14196. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Kwak, K.; Asbury, J.; Chen, X.; Piletic, I.R.; Fayer, M.D. Ultrafast dynamics of solute-solvent complexation observed at thermal equilibrium in real time. Science 2005, 309, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Senes, A.; Cristian, L.; DeGrado, W.F.; Hochstrasser, R.M. Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer. Proc. Natl. Acad. Sci. USA 2006, 103, 16740–16745. [Google Scholar] [CrossRef] [PubMed]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.-K.; Mancal, T.; Cheng, Y.-C.; Blankenship, R.E.; Fleming, G.R. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Bauman, J.D.; Das, K.; Remorino, A.; Arnold, E.; Hochstrasser, R.M. Two-dimensional infrared spectra reveal relaxation of the nonnucleoside inhibitor TMC278 complexed with the HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2008, 105, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Kurosawa, M. Femtosecond time-resolved Raman spectroscopy using stimulated Raman scattering. Phys. Rev. A 1999, 61, 013808. [Google Scholar] [CrossRef]
- McCamant, D.W.; Kukura, P.; Yoon, S.; Mathies, R.A. Femtosecond broadband stimulated Raman spectroscopy: Apparatus and methods. Rev. Sci. Instrum. 2004, 75, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Frontiera, R.R.; Fang, C.; Dasgupta, J.; Mathies, R.A. Probing structural evolution along multidimensional reaction coordinates with femtosecond stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Han, F.; Smith, C.; Fang, C. Ultrafast conformational dynamics of pyranine during excited state proton transfer in aqueous solution revealed by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10535–10550. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, J.; Frontiera, R.R.; Fang, C.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin, Germany, 2013; pp. 745–759. [Google Scholar]
- Han, F.; Liu, W.; Fang, C. Excited-state proton transfer of photoexcited pyranine in water observed by femtosecond stimulated Raman spectroscopy. Chem. Phys. 2013, 422, 204–219. [Google Scholar] [CrossRef]
- Myers, A.B.; Mathies, R.A. Resonance Raman intensities: A probe of excited-state structure and dynamics. In Biological Applications of Raman Spectroscopy; Spiro, T.G., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1987; Volume 2, pp. 1–58. [Google Scholar]
- Liu, W.; Zhu, L.; Wang, L.; Fang, C. Cascaded four-wave mixing for broadband tunable laser sideband generation. Opt. Lett. 2013, 38, 1772–1774. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, W.; Fang, C. Tunable sideband laser from cascaded four-wave mixing in thin glass for ultra-broadband femtosecond stimulated Raman spectroscopy. Appl. Phys. Lett. 2013, 103, 061110. [Google Scholar]
- Liu, W.; Zhu, L.; Fang, C. Observation of sum-frequency-generation-induced cascaded four-wave mixing using two crossing femtosecond laser pulses in a 0.1 mm beta-barium-borate crystal. Opt. Lett. 2012, 37, 3783–3785. [Google Scholar]
- Zhu, L.; Liu, W.; Wang, L.; Fang, C. Parametric amplification-assisted cascaded four-wave mixing for ultrabroad laser sideband generation in a thin transparent medium. Laser Phys. Lett. 2014, 11, 075301. [Google Scholar] [CrossRef]
- Laimgruber, S.; Schachenmayr, H.; Schmidt, B.; Zinth, W.; Gilch, P. A femtosecond stimulated Raman spectrograph for the near ultraviolet. Appl. Phys. B 2006, 85, 557–564. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, W.; Fang, C. A versatile femtosecond stimulated Raman spectroscopy setup with tunable pulses in the visible to near infrared. Appl. Phys. Lett. 2014, 105, 041106. [Google Scholar]
- Raoult, F.; Boscheron, A.C.L.; Husson, D.; Sauteret, C.; Modena, A.; Malka, V.; Dorchies, F.; Migus, A. Efficient generation of narrow-bandwidth picosecond pulses by frequency doubling of femtosecond chirped pulses. Opt. Lett. 1998, 23, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Mendonca, J.T.; Dos Santos, A. Cascaded highly nondegenerate four-wave-mixing phenomenon in transparent isotropic condensed media. Opt. Lett. 2000, 25, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kobayashi, T. Cascaded four-wave mixing and multicolored arrays generation in a sapphire plate by using two crossing beams of femtosecond laser. Opt. Express 2008, 16, 22119–22125. [Google Scholar] [CrossRef] [PubMed]
- Weigand, R.; Mendonca, J.T.; Crespo, H.M. Cascaded nondegenerate four-wave-mixing technique for high-power single-cycle pulse synthesis in the visible and ultraviolet ranges. Phys. Rev. A 2009, 79, 063838. [Google Scholar] [CrossRef]
- Shitamichi, O.; Kida, Y.; Imasaka, T. Chirped-pulse four-wave Raman mixing in molecular hydrogen. Appl. Phys. B 2014, 117, 723–730. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kukura, P.; McCamant, D.W.; Yoon, S.; Wandschneider, D.B.; Mathies, R.A. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.; Dasgupta, J.; Mathies, R.A. Femtosecond time-resolved stimulated Raman reveals the birth of bacteriorhodopsin’s J and K intermediates. J. Am. Chem. Soc. 2009, 131, 7592–7597. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, J.; Frontiera, R.R.; Taylor, K.C.; Lagarias, J.C.; Mathies, R.A. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Frontiera, R.R.; Dasgupta, J.; Mathies, R.A. Probing interfacial electron transfer in Coumarin 343 sensitized TiO2 nanoparticles with femtosecond stimulated Raman. J. Am. Chem. Soc. 2009, 131, 15630–15632. [Google Scholar] [CrossRef] [PubMed]
- Smeigh, A.L.; Creelman, M.; Mathies, R.A.; McCusker, J.K. Femtosecond time-resolved optical and Raman spectroscopy of photoinduced spin crossover: Temporal resolution of low-to-high spin optical switching. J. Am. Chem. Soc. 2008, 130, 14105–14107. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, H.; Takeuchi, S.; Tahara, T. Ultrafast structural evolution of photoactive yellow protein chromophore revealed by ultraviolet resonance femtosecond stimulated Raman spectroscopy. J. Phys. Chem. Lett. 2012, 3, 2025–2029. [Google Scholar] [CrossRef]
- Creelman, M.; Kumauchi, M.; Hoff, W.D.; Mathies, R.A. Chromophore dynamics in the PYP photocycle from femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2014, 118, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Oscar, B.G.; Liu, W.; Zhao, Y.; Tang, L.; Wang, Y.; Campbell, R.E.; Fang, C. Excited-state structural dynamics of a dual-emission calmodulin-green fluorescent protein sensor for calcium ion imaging. Proc. Natl. Acad. Sci. USA 2014, 111, 10191–10196. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Liu, W.; Wang, Y.; Zhao, Y.; Oscar, B.G.; Campbell, R.E.; Fang, C. Unraveling ultrafast photoinduced proton transfer dynamics in a fluorescent protein biosensor for Ca2+ imaging. Chem. Eur. J. 2015, 21, 6481–6490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, L.; Liu, W.; Zhao, Y.; Oscar, B.G.; Campbell, R.E.; Fang, C. Excited state structural events of a dual-emission fluorescent protein biosensor for Ca2+ imaging studied by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2015, 119, 2204–2218. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V.; Nifosì, R. Ab initio molecular dynamics of the green fluorescent protein (GFP) chromophore: An insight into the photoinduced dynamics of green fluorescent proteins. J. Phys. Chem. B 2001, 105, 5797–5803. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Chang, I.-Y.; Wills, L.A.; Zakharov, L.N.; Boettcher, S.W.; Cheong, P.H.-Y.; Fang, C.; Keszler, D.A. Electrolytic synthesis of aqueous aluminum nanoclusters and in situ characterization by femtosecond Raman spectroscopy & computations. Proc. Natl. Acad. Sci. USA 2013, 110, 18397–18401. [Google Scholar] [CrossRef] [PubMed]
- Frontiera, R.R.; Shim, S.; Mathies, R.A. Origin of negative and dispersive features in anti-Stokes and resonance femtosecond stimulated Raman spectroscopy. J. Chem. Phys. 2008, 129, 064507. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Heller, E.J. Time-dependent theory of Raman scattering. J. Chem. Phys. 1979, 71, 4777–4788. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Zhang, D.; McCamant, D.W.; Kukura, P.; Mathies, R.A. Theory of femtosecond stimulated Raman spectroscopy. J. Chem. Phys. 2004, 121, 3632–3642. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.; Stuart, C.M.; Mathies, R.A. Resonance Raman cross-sections and vibronic analysis of Rhodamine 6G from broadband stimulated Raman spectroscopy. ChemPhysChem 2008, 9, 697–699. [Google Scholar] [CrossRef] [PubMed]
- McHale, J.L. Molecular Spectroscopy; Prentice-Hall: Upper Saddle River, NJ, USA, 1999. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Liu, W.; Wang, Y.; Fang, C. Sum-Frequency-Generation-Based Laser Sidebands for Tunable Femtosecond Raman Spectroscopy in the Ultraviolet. Appl. Sci. 2015, 5, 48-61. https://doi.org/10.3390/app5020048
Zhu L, Liu W, Wang Y, Fang C. Sum-Frequency-Generation-Based Laser Sidebands for Tunable Femtosecond Raman Spectroscopy in the Ultraviolet. Applied Sciences. 2015; 5(2):48-61. https://doi.org/10.3390/app5020048
Chicago/Turabian StyleZhu, Liangdong, Weimin Liu, Yanli Wang, and Chong Fang. 2015. "Sum-Frequency-Generation-Based Laser Sidebands for Tunable Femtosecond Raman Spectroscopy in the Ultraviolet" Applied Sciences 5, no. 2: 48-61. https://doi.org/10.3390/app5020048
APA StyleZhu, L., Liu, W., Wang, Y., & Fang, C. (2015). Sum-Frequency-Generation-Based Laser Sidebands for Tunable Femtosecond Raman Spectroscopy in the Ultraviolet. Applied Sciences, 5(2), 48-61. https://doi.org/10.3390/app5020048