Plasmonic Physics of 2D Crystalline Materials

Abstract
:1. Introduction
- (a)
- Their momentum is larger than the light momentum with the same energy [4];
- (b)
- (c)
- They illustrate higher levels of confinement ( in the normal case) [4];
- (d)
- They have a longer lifetime and propagating lengths ( fs) [7];
- (e)
- They occur in the terahertz and mid-infrared modes, which are absent in normal metals [4];
- (f)
- They can be coupled with quasiparticles (for instance, generating plasmarons) [8];
- (g)
- (h)
- (k)
- At long wavelength limits, they behave like , which is proportional to the charge career as [4].
2. Theoretical Framework
2.1. Density Functional Theory
Computational Method
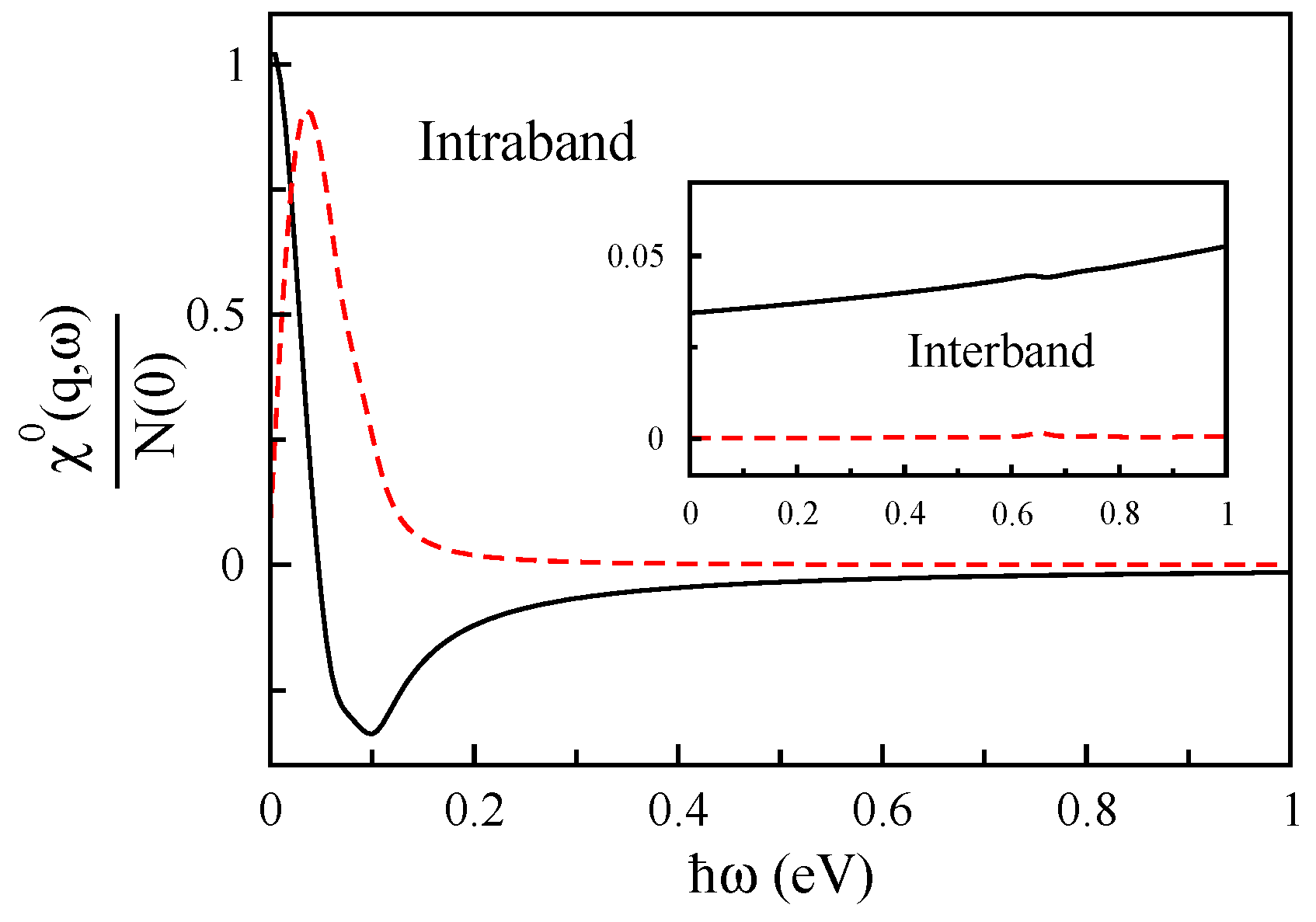
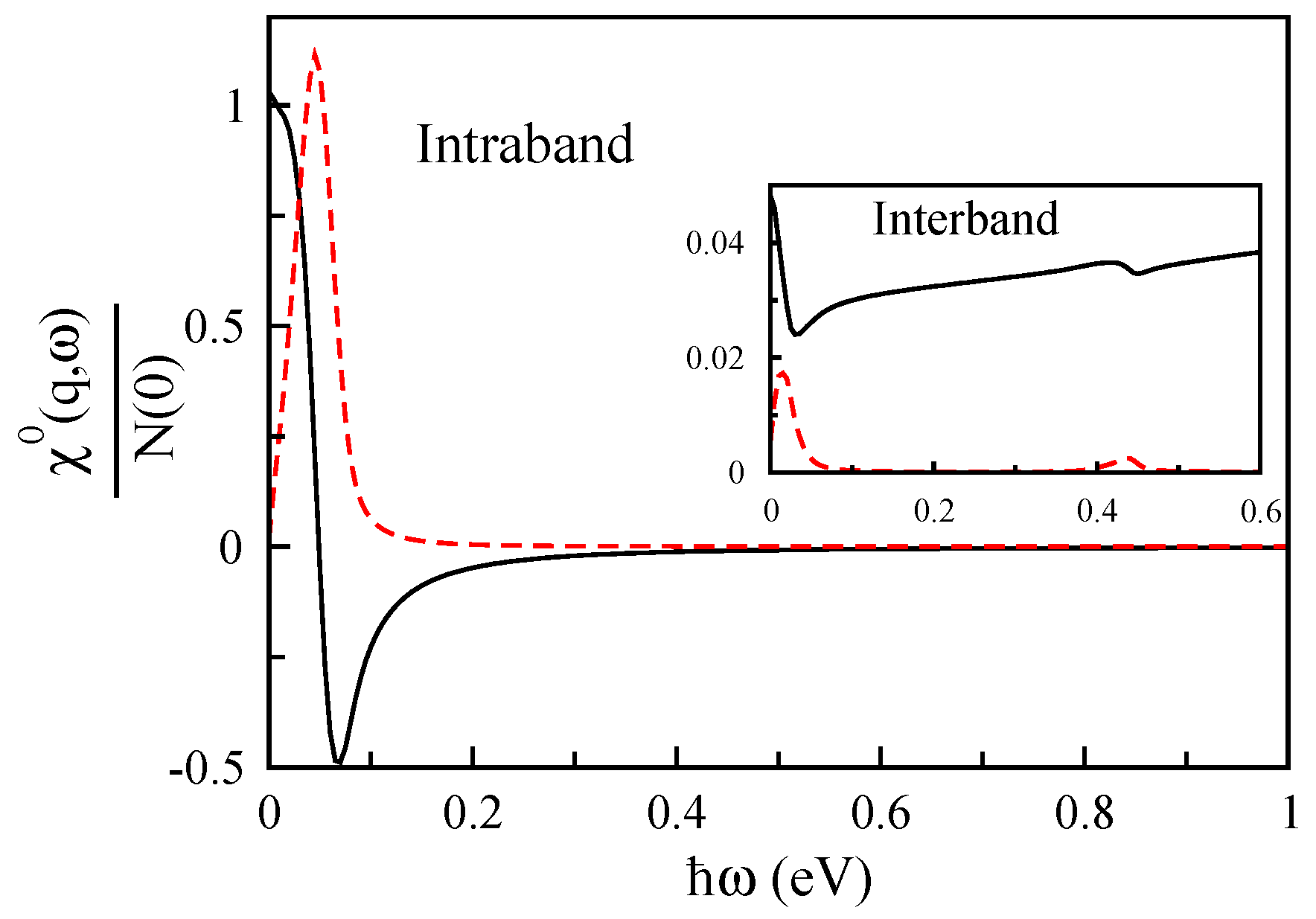
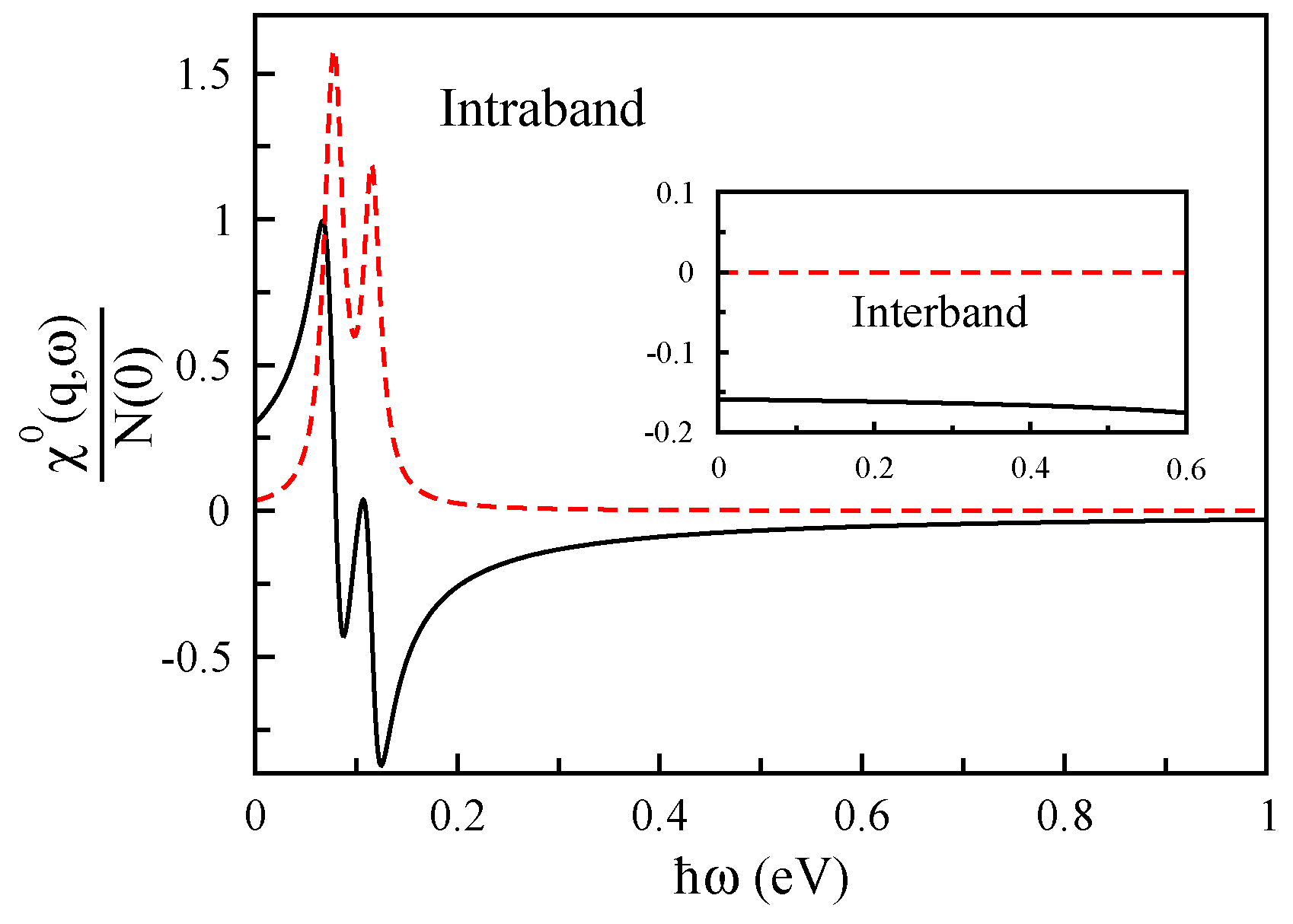
2.2. Density-Density Response Function
3. Result and Discussion
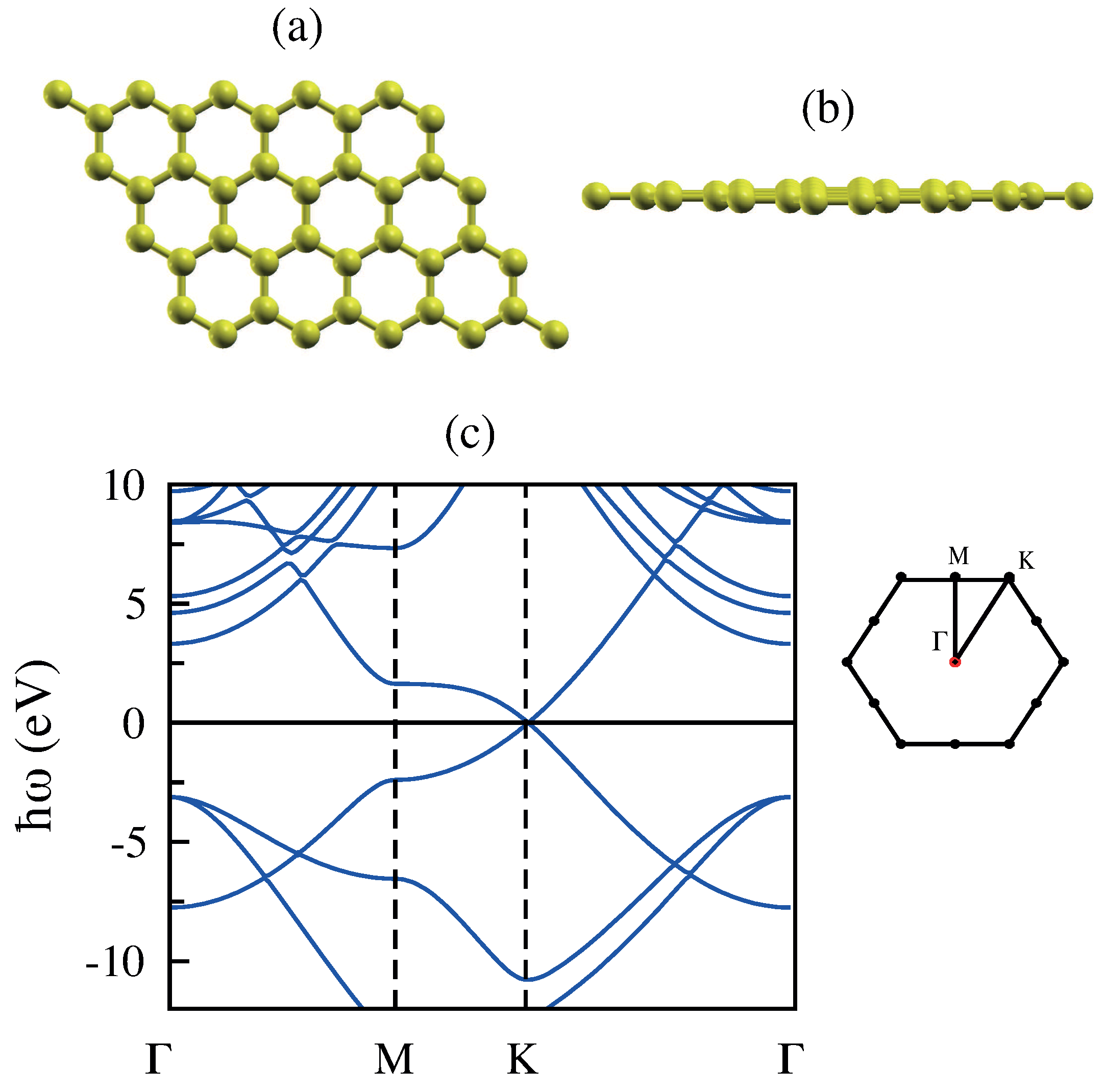
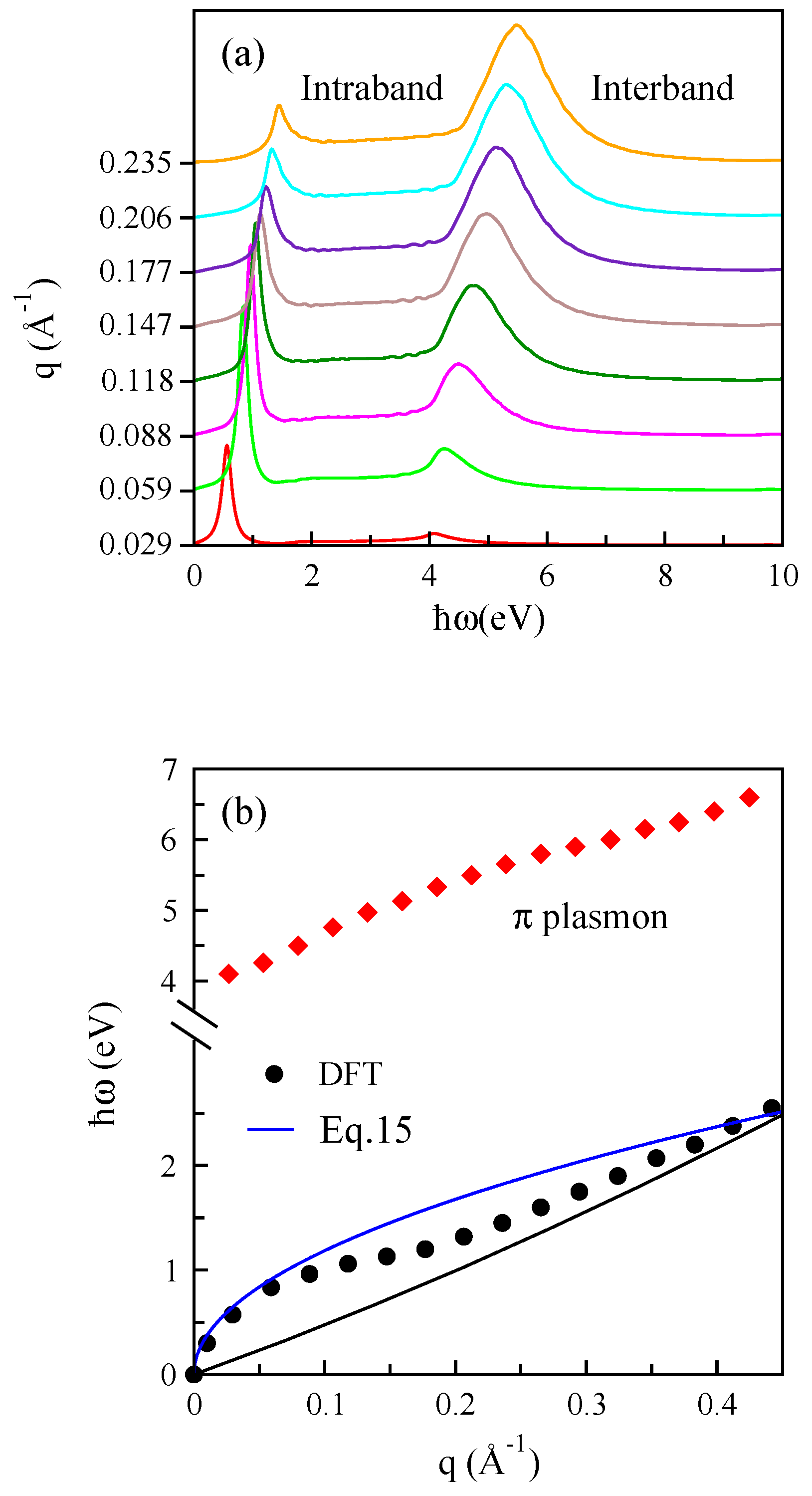
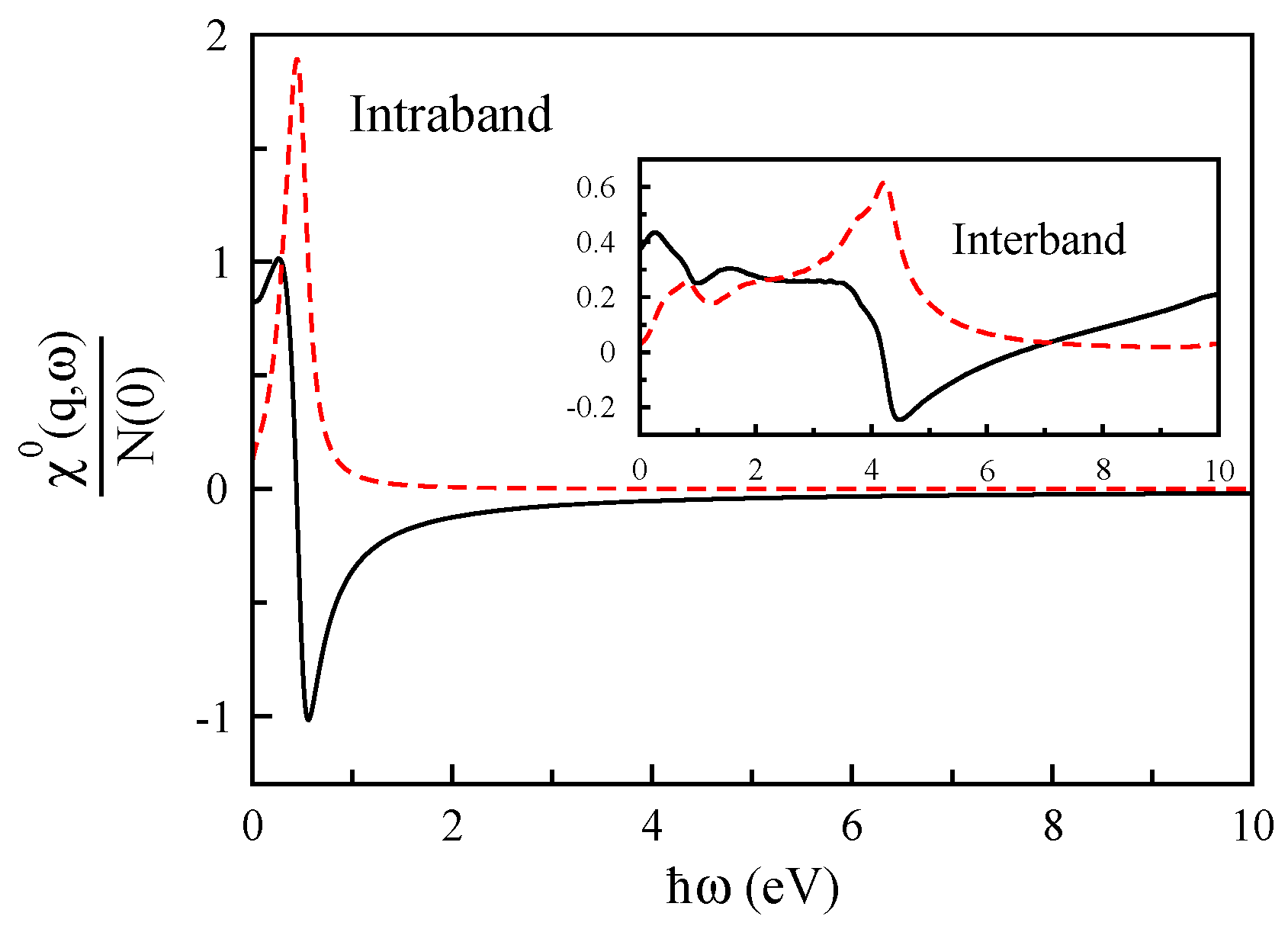
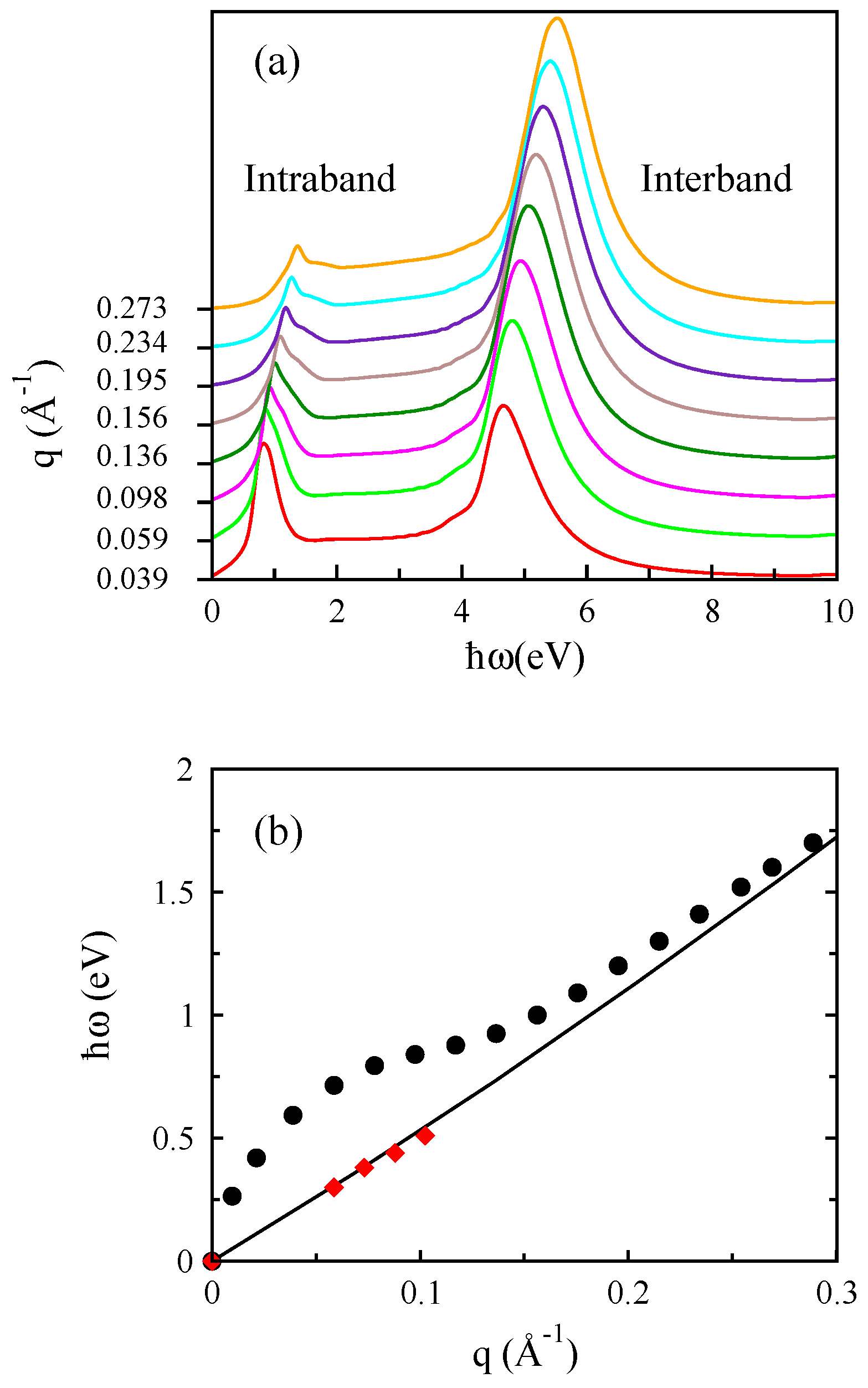
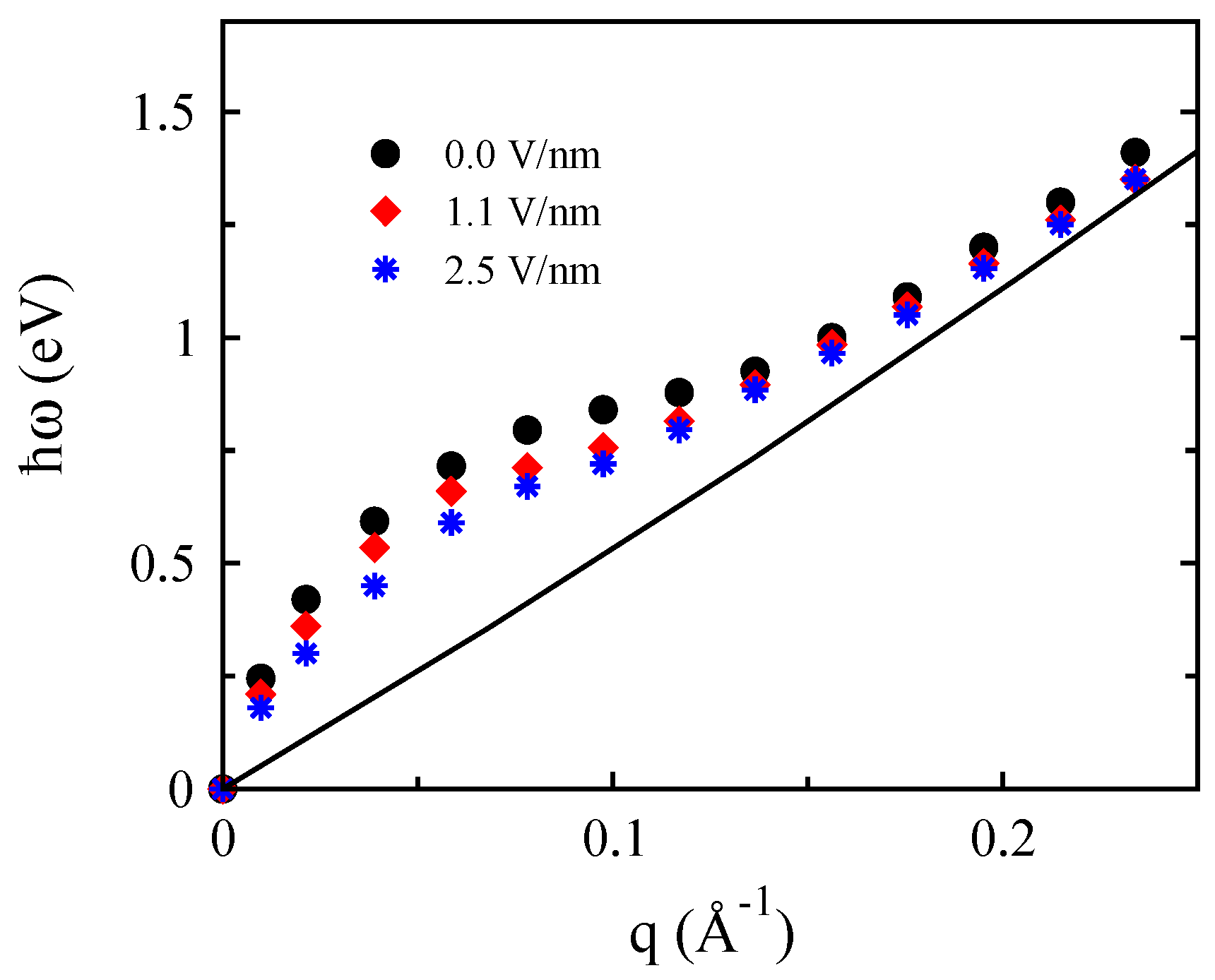
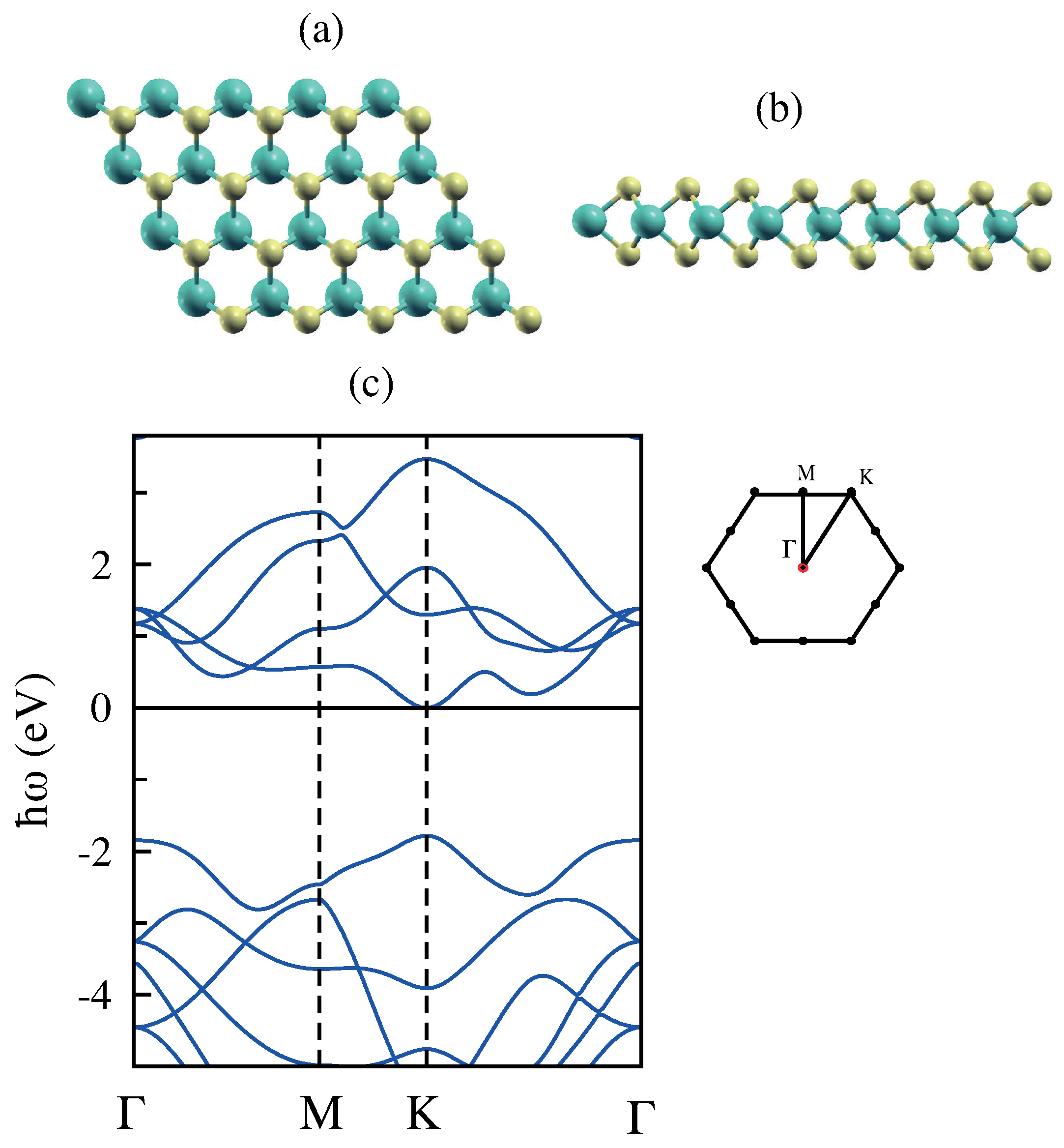
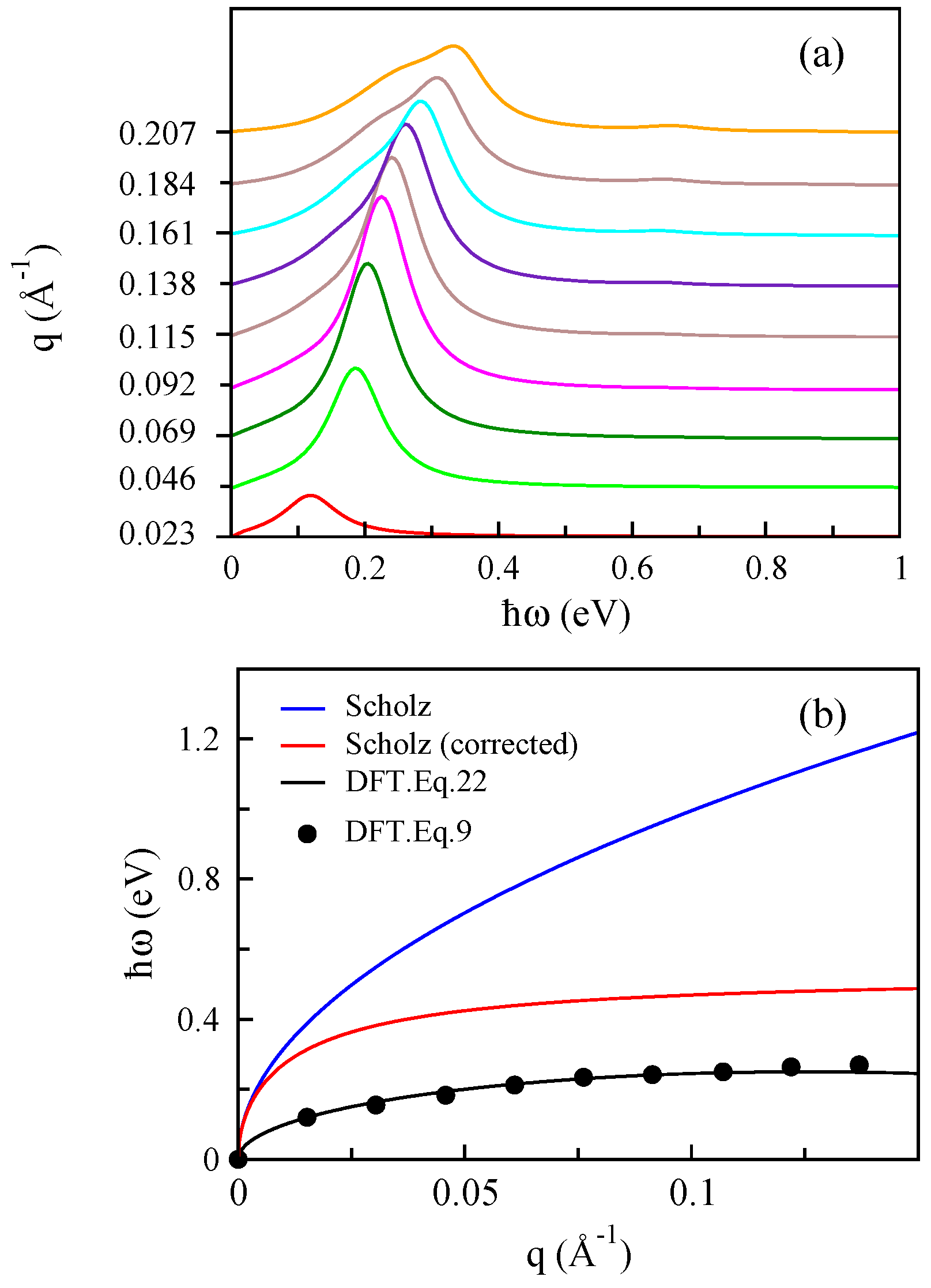
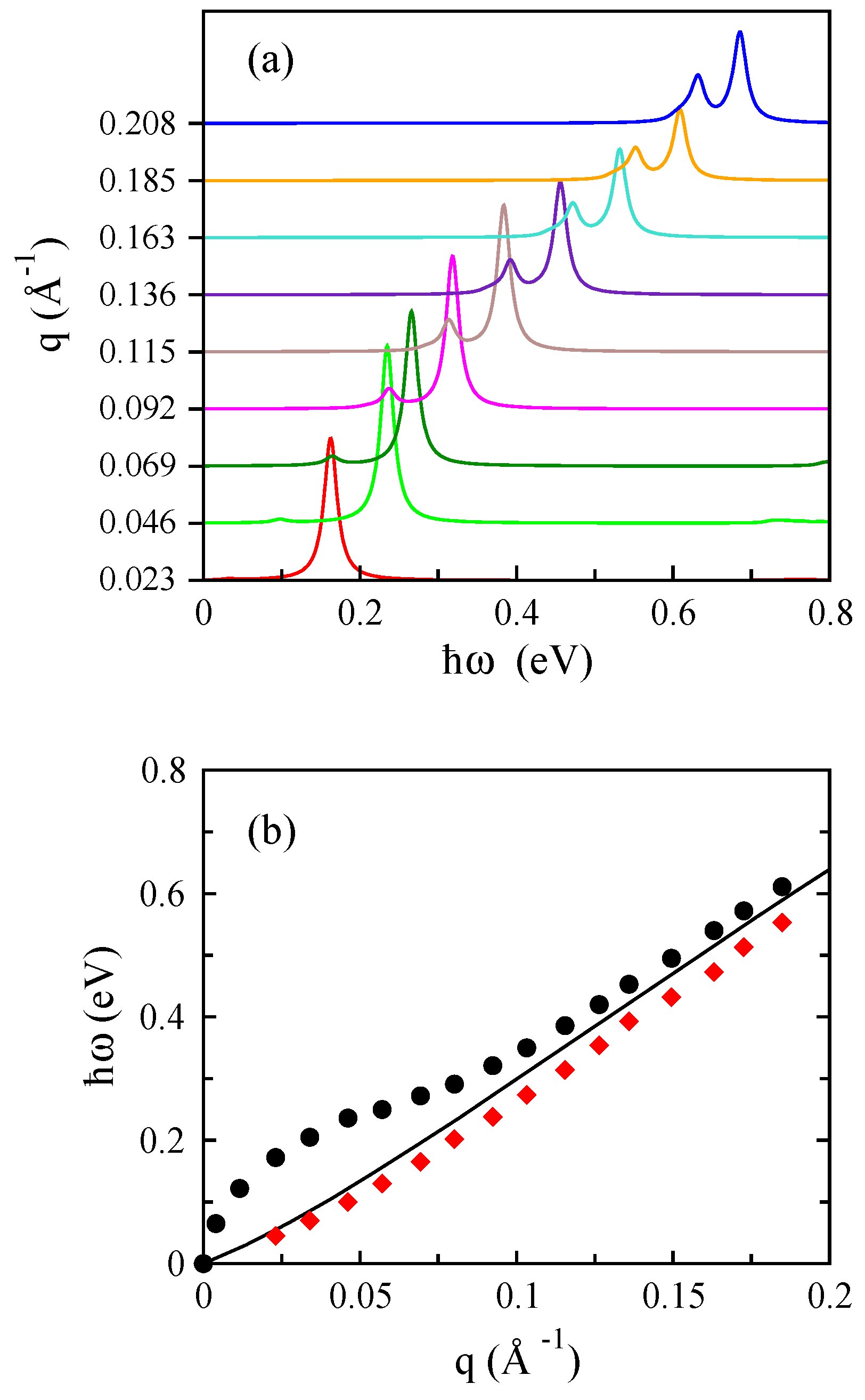
3.1. Monolayer Graphene
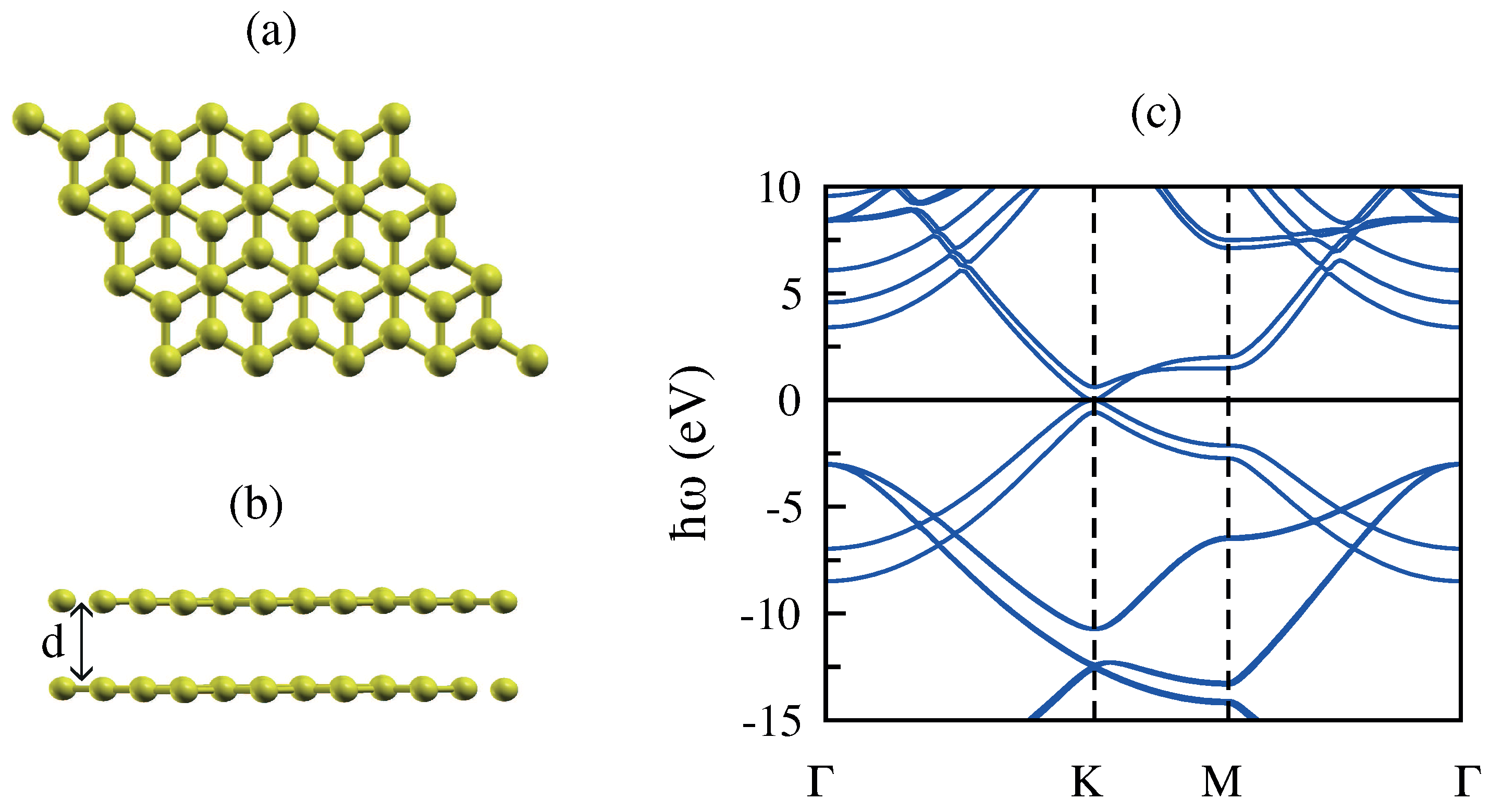
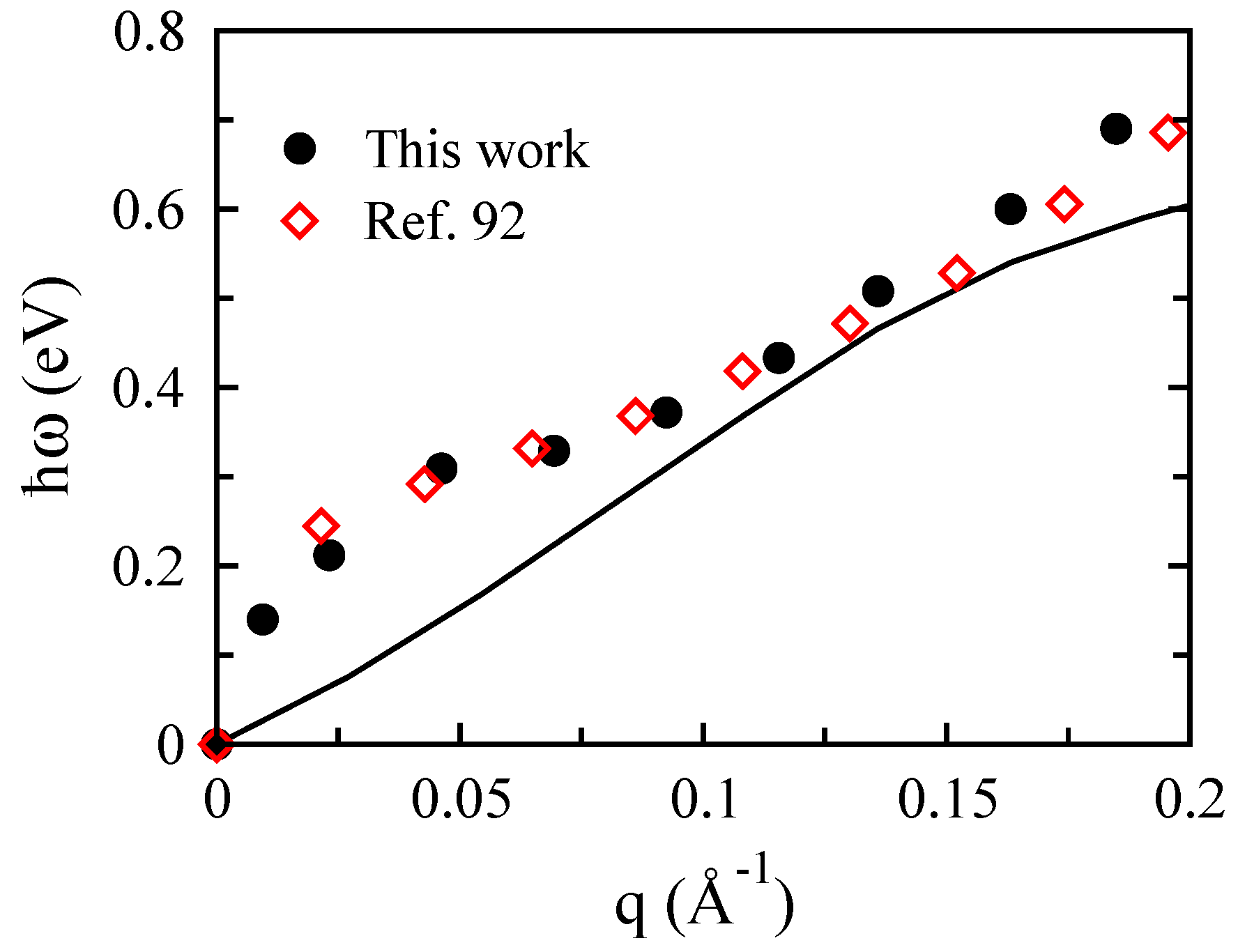
3.2. Bilayer Graphene
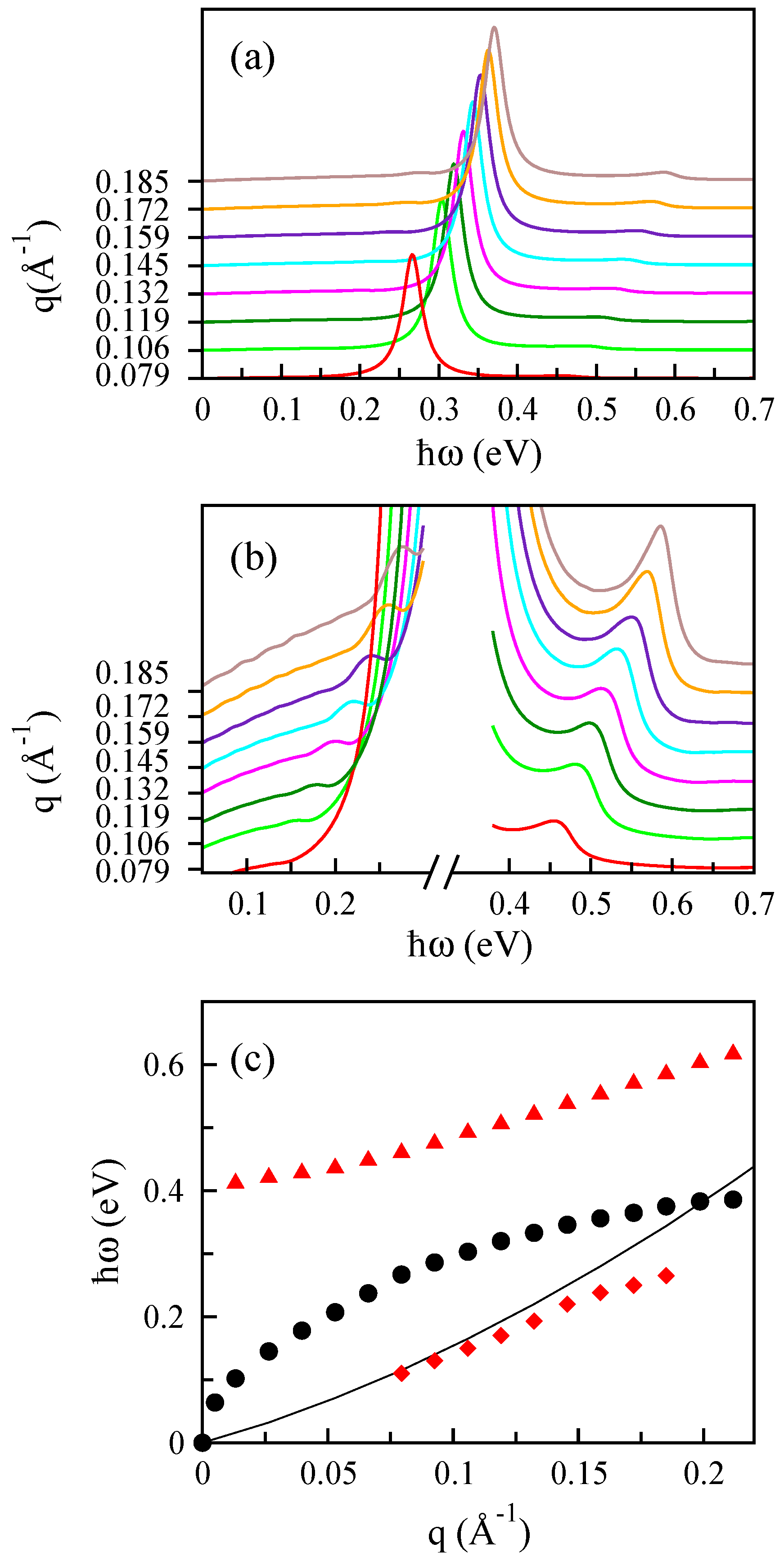
3.3. Monolayer MoS
3.4. Bilayer MoS
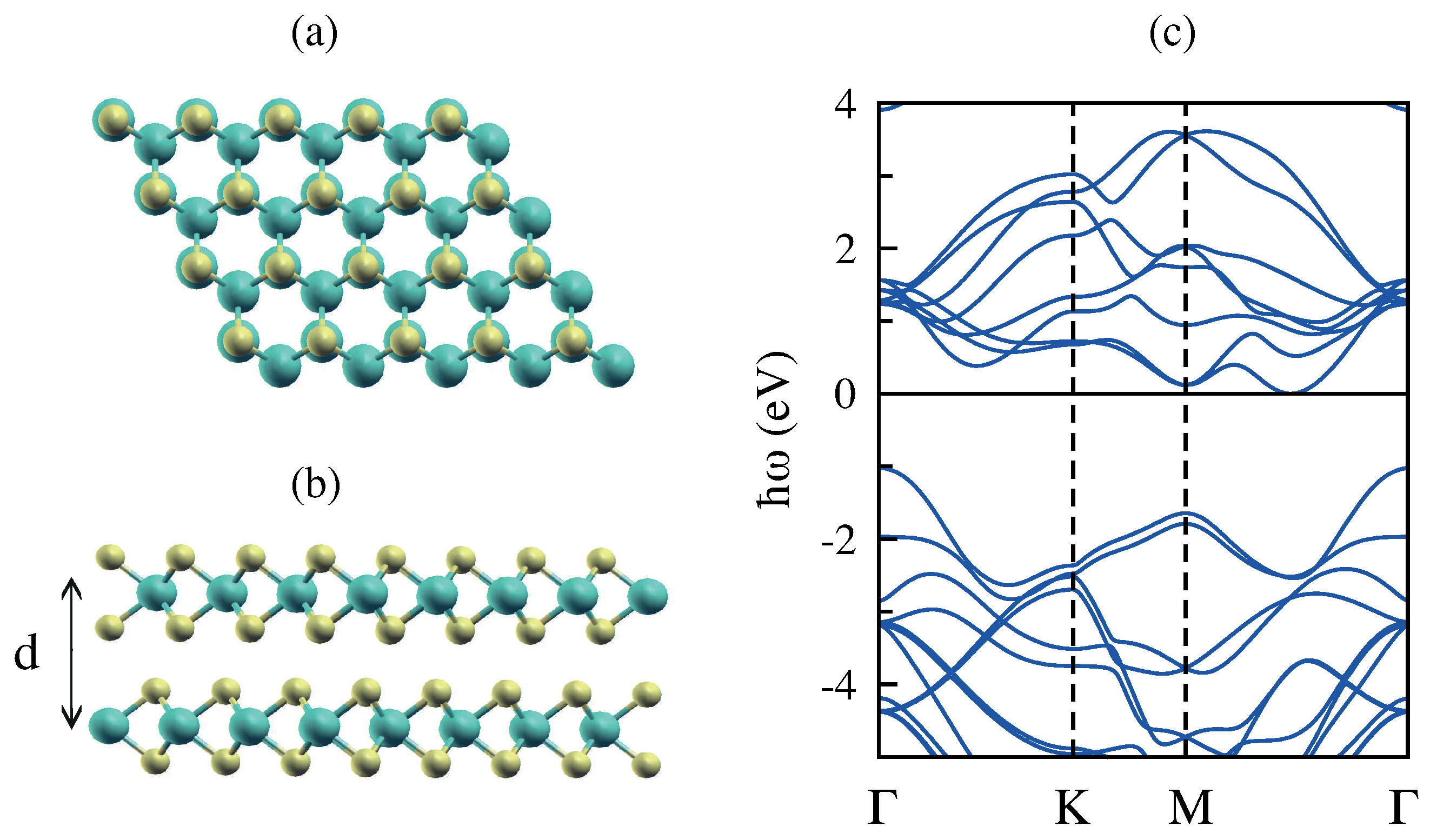
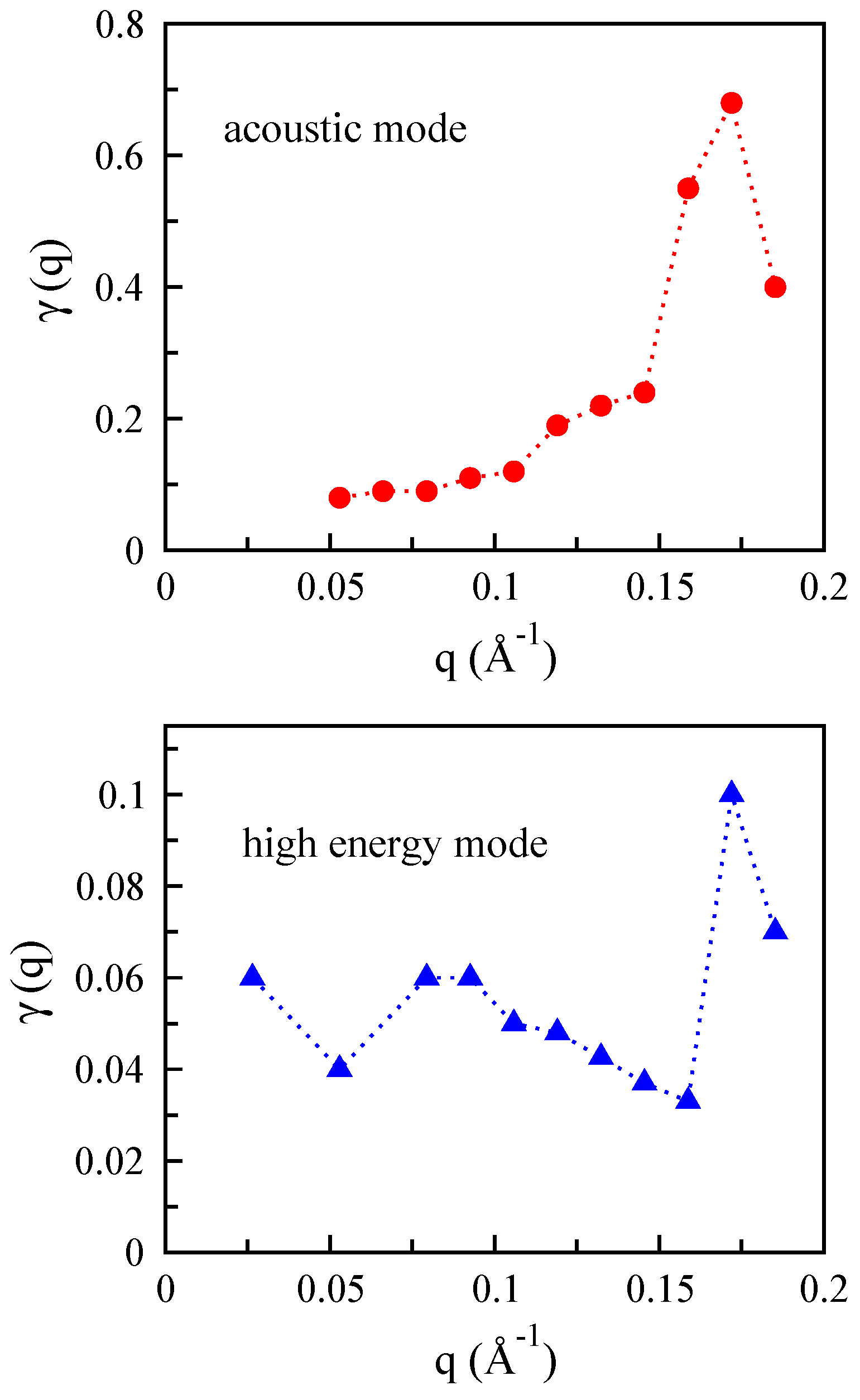
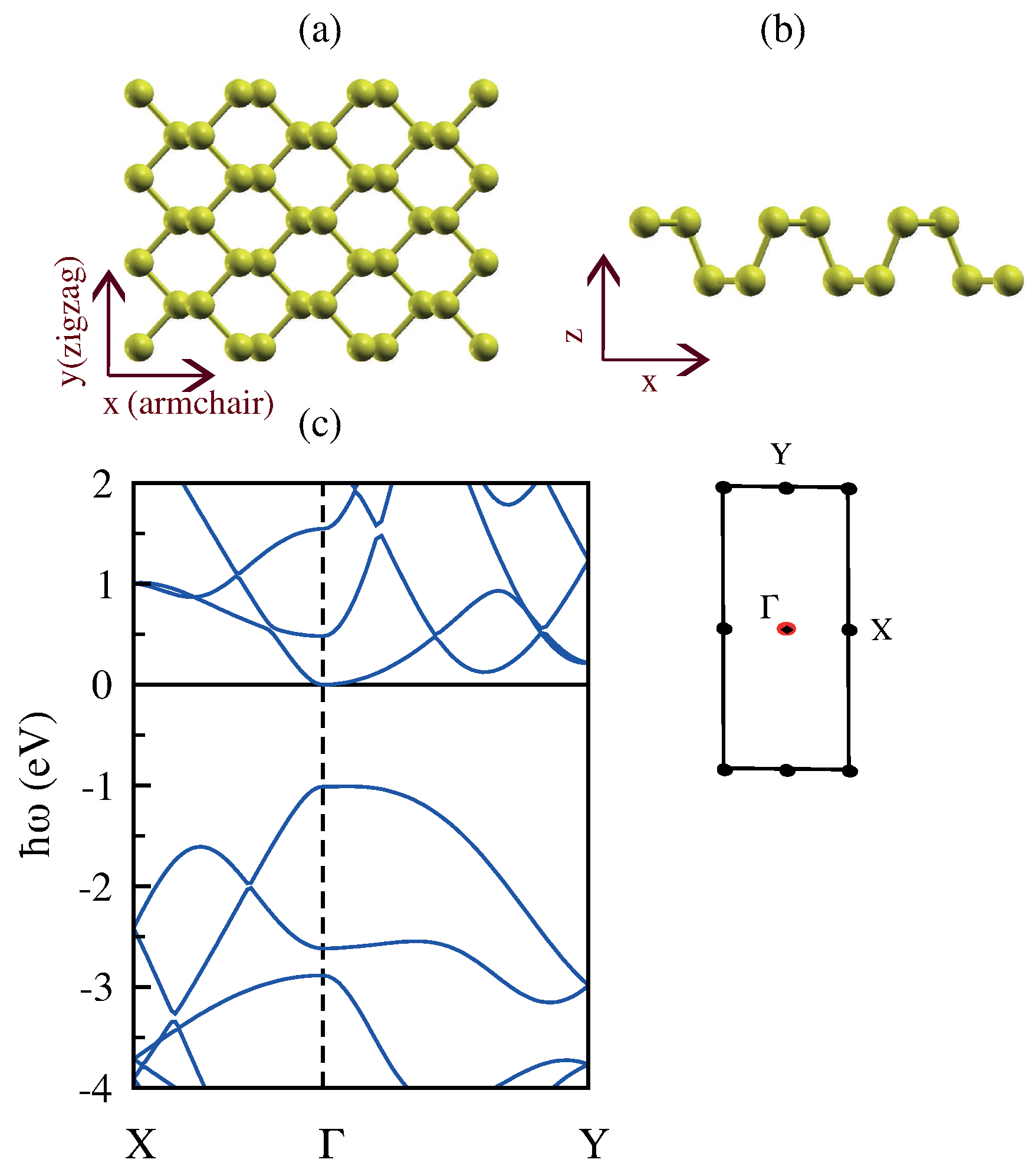
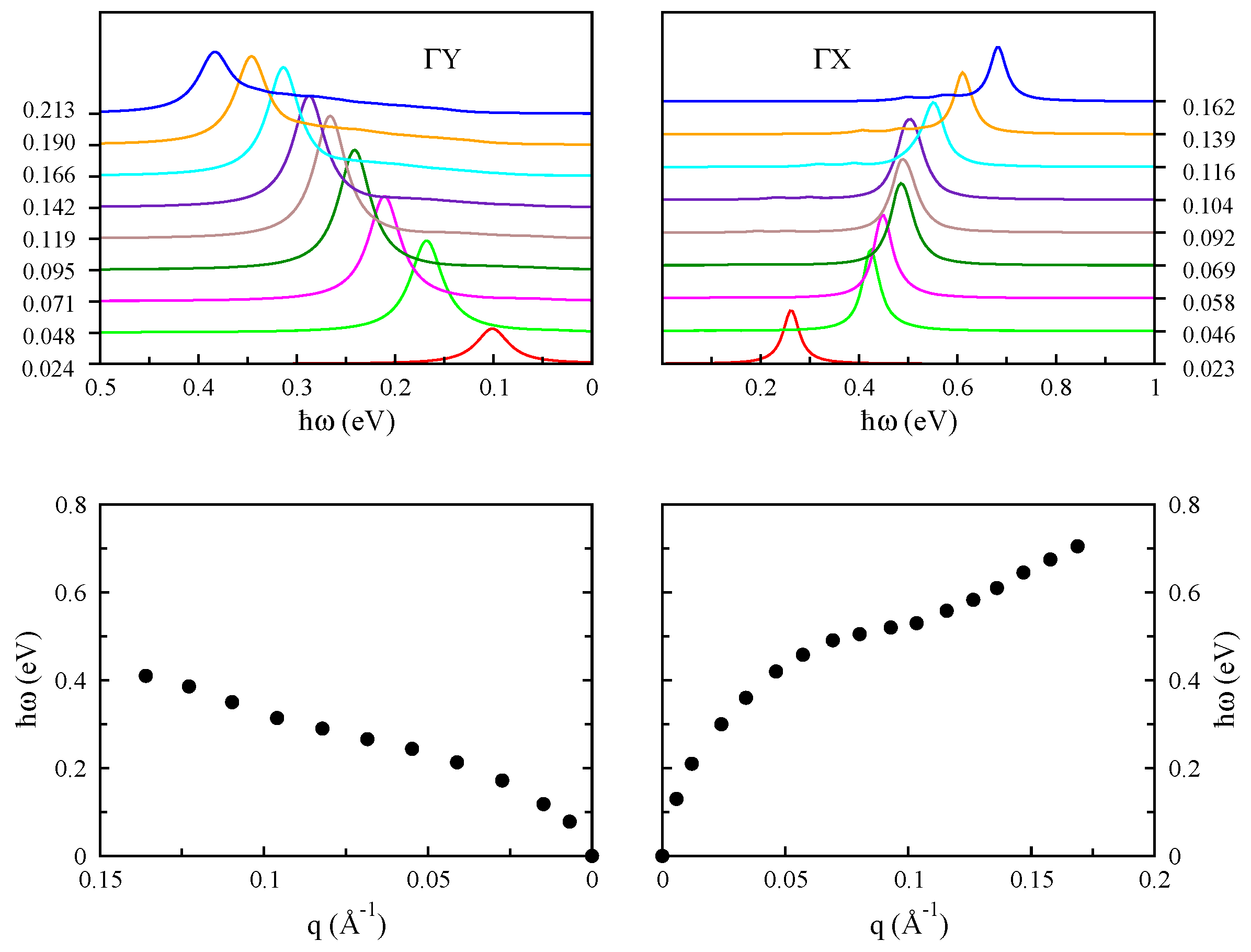
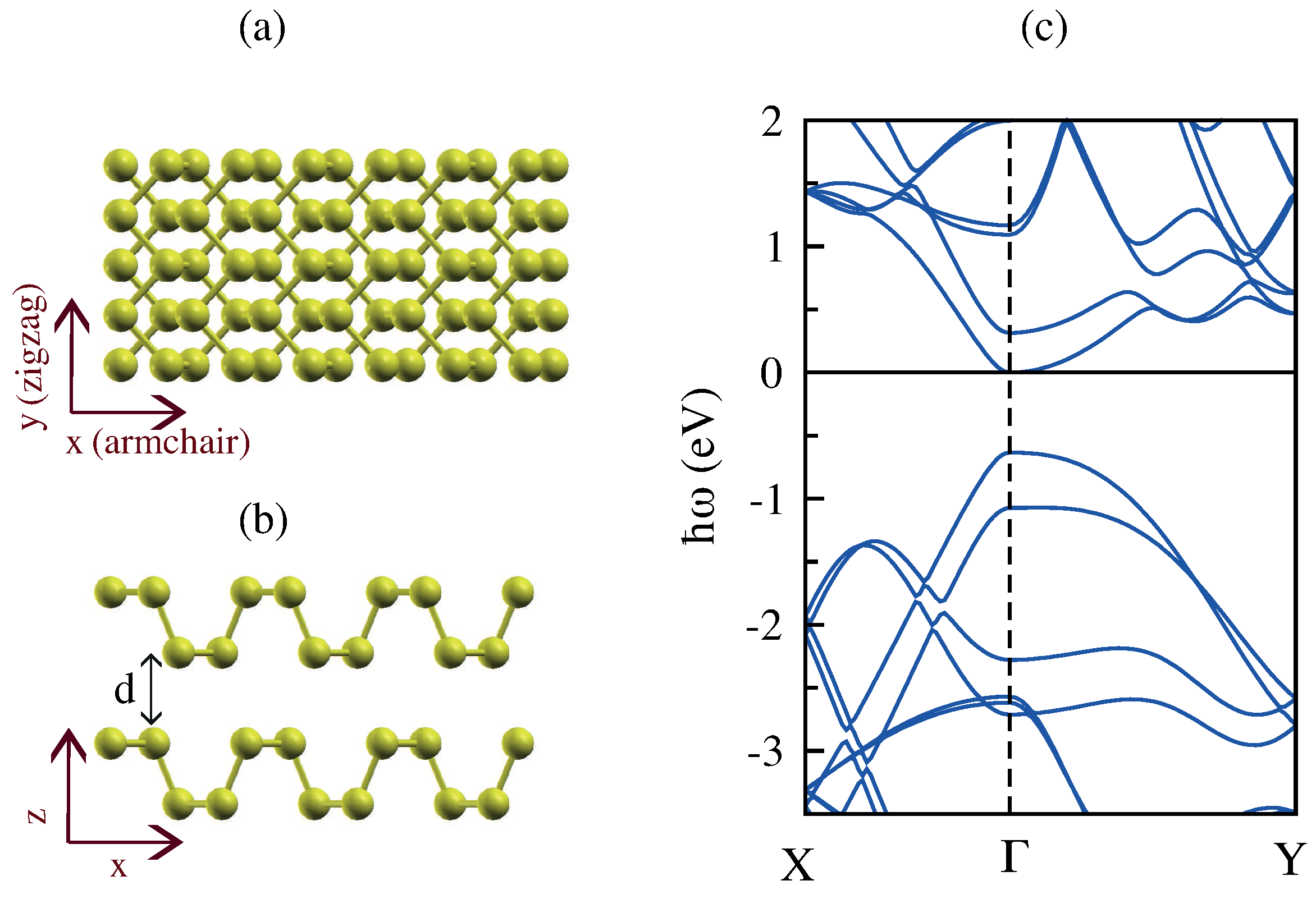
3.5. Monolayer Phosphorene
3.6. Bilayer Phosphorene
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bohm, D.; Pines, D. A Collective Description of Electron Interactions: III. Coulomb Interactions in a Degenerate Electron Gas. Phys. Rev. 1953, 92, 609. [Google Scholar] [CrossRef]
- Landau, L. On the vibrations of the electronic plasma. J. Phys. (USSR) 1946, 10, 25. [Google Scholar]
- Ritchie, R.H. Plasma Losses by Fast Electrons in Thin Films. Phys. Rev. 1957, 106, 874. [Google Scholar] [CrossRef]
- Koppens, F.H.L.; Chang, D.E.; de Abajo, F.J.G. Graphene Plasmonics: A Platform for Strong Light—Matter Interactions. Nano Lett. 2011, 11, 3370–3377. [Google Scholar] [CrossRef] [PubMed]
- Fer, Z.; Rodin, A.S.; Andreev, G.O.; Bao, W.; McLeod, A.S.; Wagner, M.; Zhang, L.M.; Zhao, Z.; Thiemens, M.; Dominguez, G.; et al. Gate-tuning of graphene plasmons reveald by infrared nano-imaging. Nature 2012, 487, 82–85. [Google Scholar] [CrossRef]
- Chen, J.; Badioli, M.; Alonso-González, P.; Thongrattanasiri, S.; Huth, F.; Osmond, J.; Spasenović, M.; Centeno, A.; Pesquera, A.; Godignon, P.; et al. Optical nano-imaging of gate-tunable graphene plasmons. Nature 2012, 487, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Lundeberg, M.; Gao, Y.; Asgari, R.; Tan, C.; Duppen, B.V.; Autore, M.; Alonso-Gonzalez, P.; Woessner, A.; Watanabe, K.; Taniguchi, T.; et al. Tuning quantum nonlocal effects in graphene plasmonics. Science 2017, 357, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, A.; Speck, F.; Seyller, T.; Horn, K.; Polini, M.; Asgari, R.; MacDonald, A.H.; Rotenberg, E. Observation of Plasmarons in Quasi-Free-Standing Doped Graphene. Science 2010, 328, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Kasry, A.; Ardakani, A.A.; Tulevski, G.S.; Menges, B.; Copel, M.; Vyklicky, L. Highly efficient fluorescence quenching with graphene. J. Phys. Chem. C 2012, 116, 2858–2862. [Google Scholar] [CrossRef]
- Gaudreau, L.; Tielrooij, K.J.; Prawiroatmodjo, G.E.D.K.; Osmond, J.; de Abajo, F.J.G.; Koppens, F.H.L. Universal distance-scaling of nonradiative enegy transfer to graphener. Nano Lett. 2013, 13, 2030–2035. [Google Scholar] [CrossRef] [PubMed]
- Principi, A.; Carrega, M.; Asgari, R.; Pellegrini, V.; Polini, M. Plasmons and Coulomb Drag in Dirac/Schroedinger Hybrid Electron Systems. Phys. Rev. B 2012, 86, 085421. [Google Scholar] [CrossRef]
- Faridi, A.; Asgari, R. Plasmons at the LaAlO3/SrTiO3 interface and Graphene-LaAlO3/SrTiO3 double layer. Phys. Rev. B 2017, 95, 165419. [Google Scholar] [CrossRef]
- Loudon, R. The propagation of electromagnetic energy through an absorbing dielectric. J. Phys. A 1970, 3, 233. [Google Scholar] [CrossRef]
- Maier, S.A. Plasmonics: Fundamentals and Applications; Springer: Boston, MA, USA, 2007. [Google Scholar]
- Wang, Y.; Plummer, E.W.; Kempa, K. Foundations of Plasmonics. Adv. Phys. 2011, 60, 799–898. [Google Scholar] [CrossRef]
- Garcia de Abajo, F.J. Graphene Plasmonics: Challenges and Opportunities. ACS Photonics 2014, 1, 135–152. [Google Scholar] [CrossRef]
- Jablan, M.; Sljačić, M.; Buljan, H. Plasmons in Graphene: Fundamental Properties and Potential Applications. Proc. IEEE 2013, 101, 1689–1704. [Google Scholar] [CrossRef]
- Yan, H.; Low, T.; Zhu, W.; Wu, Y.; Freitag, M.; Li, X.; Guinea, F.; Avouris, P.; Xia, F. Damping pathways of mid-infrared plasmons in graphene nanostructures. Nat. Photonics 2013, 7, 394–399. [Google Scholar] [CrossRef]
- Goncalves, P.A.D.; Peres, N.M.R. An Introduction to Graphene Plasmonics; World Scientific Publishing: Singapore, 2016. [Google Scholar]
- Tame, M.S.; McEnery, K.R.; Özdemir, S.K.; Lee, J.; Maier, S.A.; Kim, M.S. Quantum plasmonics. Nat. Phys. 2013, 9, 329–340. [Google Scholar] [CrossRef]
- Sholl, D.S.; Steckel, J.A. Density Functional Theory: A Practical Introduction; Wiley-Interscience: New York, NY, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford: New York, NY, USA, 1989. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Moroni, E.G.; Kresse, G.; Hafner, J.; Furthmüller, J. Ultrasoft pseudopotentials applied to magnetic Fe, Co, and Ni: From atoms to solids. Phys. Rev. B 1997, 56, 15629. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef]
- Chadi, D.J.; Cohen, M.L. Special Points in the Brillouin Zone. Phys. Rev. B 1973, 8, 5747. [Google Scholar] [CrossRef]
- Giuliani, G.F.; Vignale, G. Quantum Theory of the Electron Liquid; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Adler, S.L. Quantum theory of the dielectric constant in real solids. Phys. Rev. 1962, 126, 413. [Google Scholar] [CrossRef]
- Petersilka, M.; Gossmann, U.J.; Gross, E.K.U. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212. [Google Scholar] [CrossRef] [PubMed]
- Pisarra, M.; Sindona, A.; Gravina, M.; Silkin, V.M.; Pitarke, J.M. Dielectric screening and plasmon resonances in bilayer graphene. Phys. Rev. B 2016, 93, 035440. [Google Scholar] [CrossRef]
- Gomez, C.V.; Pisarra, M.; Gravina, M.; Pitarke, J.M.; Sindona, A. Plasmon Modes of Graphene Nanoribbons with Periodic Planar Arrangements. Phys. Rev. Lett 2016, 117, 116801. [Google Scholar] [CrossRef] [PubMed]
- Kramberger, C.; Hambach, R.; Giorgetti, C.; Rümmeli, M.H.; Knupfer, M.; Fink, J.; Büchner, B.; Reining, L.; Einarsson, E.; Maruyama, S.; et al. Linear plasmon dispersion in single-wall carbon nanotubes and the collective excitation spectrum of graphene. Phys. Rev. Lett 2008, 100, 196803. [Google Scholar] [CrossRef] [PubMed]
- Rostami, H.; Asgari, R. Electronic ground-state properties of strained graphene. Phys. Rev. B 2012, 86, 155435. [Google Scholar] [CrossRef]
- Polini, M.; Tomadin, A.; Asgari, R.; MacDonald, A.H. Density functional theory of graphene sheets. Phys. Rev. B 2008, 78, 115426. [Google Scholar] [CrossRef]
- Neto, A.H.C.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109. [Google Scholar] [CrossRef]
- Despoja, V.; Dekanić, K.; Šunjić, M.; Marušić, L. Ab initio study of energy loss and wake potential in the vicinity of a graphene monolayer. Phys. Rev. B 2012, 86, 165419. [Google Scholar] [CrossRef]
- Yan, J.; Thygesen, K.S.; Jacobsen, K.W. Nonlocal Screening of Plasmons in Graphene by Semiconducting and Metallic Substrates: First-Principles Calculations. Phys. Rev. Lett. 2011, 106, 146803. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, B.; Stauber, T.; Sols, F.; Guinea, F. Dynamical polarization of graphene at finite doping. New J. Phys. 2006, 8, 318. [Google Scholar] [CrossRef]
- Jablan, M.; Buljan, H.; Soljačić, M. Plasmonics in graphene at infrared frequencies. Phys. Rev. B 2009, 80, 245435. [Google Scholar] [CrossRef]
- Wachsmuth, P.; Hambach, R.; Kinyanjui, M.K.; Guzzo, M.; Benner, G.; Kaiser, U. High-energy collective electronic excitations in free-standing single-layer graphene. Phys. Rev. B 2013, 88, 075433. [Google Scholar] [CrossRef]
- Gao, Y.; Yuan, Z. Anisotropic low-energy plasmon excitations in doped graphene: An ab initio study. Solid State Commun. 2011, 151, 1009–1013. [Google Scholar] [CrossRef]
- Novko, D.; Despoja, V.; Šunjić, M. Changing character of electronic transitions in graphene: From single-particle excitations to plasmons. Phys. Rev. B 2015, 91, 195407. [Google Scholar] [CrossRef]
- Stauber, T. Plasmonics in Dirac systems: From graphene to topological insulators. J. Phys. Condens. Matter 2014, 26, 123201. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ren, X.; He, L. First-principles calculations and model analysis of plasmon excitations in graphene and graphene/hBN heterostructure. Phys. Rev. B 2017, 96, 165417. [Google Scholar] [CrossRef]
- Eberlein, T.; Bangert, U.; Nair, R.R.; Jones, R.; Gass, M.; Bleloch, A.L.; Novoselov, K.S.; Geim, A.; Briddon, P.R. Plasmon spectroscopy of free-standing graphene films. Phys. Rev. B 2008, 77, 233406. [Google Scholar] [CrossRef]
- Despoja, V.; Novko, D.; Dekanić, K.; Šunjić, M.; Marušić, L. Two-dimensional and π plasmon spectra in pristine and doped graphene. Phys. Rev. B 2013, 87, 075447. [Google Scholar] [CrossRef]
- McCann, E.; Koshino, M. The electronic properties of bilayer graphene. Rep. Prog. Phys. 2013, 76, 056503. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Sahu, B.; Banerjee, S.K.; MacDonald, A.H. Ab initio theory of gate induced gaps in graphene bilayers. Phys. Rev. B 2007, 75, 155115. [Google Scholar] [CrossRef]
- Ohta, T.; Bostwick, A.; Seyller, T.; Horn, K.; Rotenberg, E. Controlling the electronic structure of bilayer graphene. Science 2006, 313, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, P.; Hambach, R.; Benner, G.; Kaiser, U. Plasmon bands in multilayer graphene. Phys. Rev. B 2014, 90, 235434. [Google Scholar] [CrossRef]
- Borghi, G.; Polini, M.; Asgari, R.; MacDonald, A.H. Dynamical response functions and collective modes of bilayer graphene. Phys. Rev. B 2009, 80, 241402. [Google Scholar] [CrossRef]
- Profumo, R.E.V.; Asgari, R.; Polini, M.; MacDonald, A.H. Double-layer graphene and topological insulator thin-film plasmons. Phys. Rev. B 2012, 85, 085443. [Google Scholar] [CrossRef]
- Sarma, S.D.; Wang, E.H. Plasmons in Coupled Bilayer Structures. Phys. Rev. Lett. 1998, 81, 4216. [Google Scholar] [CrossRef]
- McCann, E. Ab initio theory of gate induced gaps in graphene bilayers. Phys. Rev. B 2006, 74, 161403. [Google Scholar] [CrossRef]
- Guinea, F.; Neto, A.H.C.; Peres, N.M.R. Electronic states and Landau levels graphene stacks. Phys. Rev. B 2005, 73, 245426. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, T.T.; Girit, C.; Hao, Z.; Martin, M.C.; Zettl, A.; Crommie, M.F.; Shen, Y.B.; Wang, F. Direct observation of a widely tunable bandgap in bilayer graphene. Nature 2009, 11, 820. [Google Scholar] [CrossRef] [PubMed]
- Skakalova, V.; Kaiser, A.B. Graphene, Properties, Preparation, Characterisation and Devices; Woodhead Publishing of Elsevier: Cambridge, UK, 2014. [Google Scholar]
- Wang, X.F.; Chakraborty, T. Coulomb screening and collective excitations in biased bilayer graphene. Phys. Rev. B 2010, 81, 081402. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Sun, Z.; Hasan, T.; Ferrari, A.C. Graphene photonics and optoelectronics. Nat. Photonics 2010, 4, 611. [Google Scholar] [CrossRef]
- Rukelj, Z.; Štrkalj, A.; Despoja, V. Optical absorption and transmission in a molybdenum disulfide monolayer. Phys. Rev. B 2016, 94, 115428. [Google Scholar] [CrossRef]
- Kadantsev, E.S.; Hawrylak, P. Electronic structure of a single MoS2 monolayer. Solid State Commun. 2012, 152, 909–913. [Google Scholar] [CrossRef]
- Kumara, A.; Ahluwalia, P.K. Electronic structure of transition metal dichalcogenides monolayers 1H-MX2 (M = Mo, W; X = S, Se, Te) from ab-initio theory: New direct band gap semiconductors. Eur. Phys. J. B 2012, 85, 186. [Google Scholar] [CrossRef]
- Rostami, H.; Roldan, R.; Cappelluti, E.; Asgari, R.; Guinea, F. Theory of strain in single-layer transition metal dichalcogenides. Phys. Rev. B 2015, 92, 195402. [Google Scholar] [CrossRef]
- Rostami, H.; Asgari, R. Valley Zeeman effect and spin-valley polarized conductance in monolayer MoS2 in a perpendicular magnetic field. Phys. Rev. B 2015, 91, 075433. [Google Scholar] [CrossRef]
- Shishkin, M.; Kresse, G. Self-consistent GW calculations for semiconductors and insulators. Phys. Rev. B 2007, 75, 235102. [Google Scholar] [CrossRef]
- Qiu, D.Y.; da Jornada, F.H.; Louie, S.G. Optical Spectrum of MoS2: Many-Body Effects and Diversity of Exciton States. Phys. Rev. Lett. 2013, 111, 216805. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.; Thygesen, K.S. Plasmons in metallic monolayer and bilayer transition metal dichalcogenides. Phys. Rev. B 2013, 88, 155128. [Google Scholar] [CrossRef]
- Scholz, A.; Stauber, T.; Schliemann, J. Plasmons and screening in a monolayer of MoS2. Phys. Rev. B 2008, 88, 035135. [Google Scholar] [CrossRef]
- Groenewald, R.E.; Rösner, M.; Schönhoff, G.; Haas, S.; Wehling, T.O. Valley plasmonics in transition metal dichalcogenides. Phys. Rev. B 2016, 93, 205145. [Google Scholar] [CrossRef]
- Cudazzo, P.; Tokatly, I.V.; Rubio, A. Dielectric screening in two-dimensional insulators: Implications for excitonic and impurity states in graphane. Phys. Rev. B 2011, 84, 085406. [Google Scholar] [CrossRef]
- Keldysh, L.V. Coulomb interaction in thin semiconductor and semimetal films. JETP Lett. 1979, 29, 658. [Google Scholar]
- Gutiérrez-Rubio, A.; Stauber, T.; Gómez-Santos, G.; Asgari, R.; Guinea, F. Orbital magnetic susceptibility of graphene and MoS2. Phys. Rev. B 2016, 93, 085133. [Google Scholar] [CrossRef]
- Rostami, H.; Asgari, R. Valley Zeeman effect and spin-valley polarized conductance in monolayer MoS2 in a perpendicular magnetic field. Phys. Rev. B 2015, 91, 075433. [Google Scholar] [CrossRef]
- Cheiwchanchamnangij, T.; Lambrecht, W.R.L. Quasiparticle band structure calculation of monolayer, bilayer, and bulk MoS2. Phys. Rev. B 2012, 85, 205302. [Google Scholar] [CrossRef]
- Debbichi, L.; Eriksson, O.; Lebegue, S. Electronic structure of two-dimensional transition metal dichalcogenide bilayers from ab initio theory. Phys. Rev. B 2014, 89, 205311. [Google Scholar] [CrossRef]
- Komsa, H.-P.; Krasheninnikov, A.V. Effects of confinement and environment on the electronic structure and exciton binding energy of MoS2 from first principles. Phys. Rev. B 2012, 86, 241201. [Google Scholar] [CrossRef]
- Ramasubramaniam, A.; Naveh, D.; Towe, E. Tunable band gaps in bilayer transition-metal dichalcogenides. Phys. Rev. B 2011, 84, 205325. [Google Scholar] [CrossRef]
- Torbatian, Z.; Asgari, R. Plasmon modes of bilayer molybdenum disulfide: A density functional study. J. Phys. Condens. Matter 2017, 29, 465701. [Google Scholar] [CrossRef] [PubMed]
- Schwierz, F. Graphene transistors. Nat. Nanotech. 2010, 5, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Rameshti, B.Z.; Ghamsari, F.G.; Asgari, R. Thermoelectric transport in monolayer phosphorene. Phys. Rev. B 2017, 95, 045422. [Google Scholar] [CrossRef]
- Wang, V.; Kawazoe, Y.; Geng, W.T. Native point defects in few-layer phosphorene. Phys. Rev. B 2015, 91, 045433. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Elahi, M.; Khaliji, K.; Tabatabaei, S.M.; Pourfath, M.; Asgari, R. Modulation of electronic and mechanical properties of phosphorene through strain. Phys. Rev. B 2015, 91, 115412. [Google Scholar] [CrossRef]
- Tran, V.; Soklaski, R.; Liang, Y.; Yang, L. Layer-controlled band gap and anisotropic excitons in few-layer black phosphorus. Phys. Rev. B 2014, 89, 235319. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.D.S.O.; Linghu, J.; Zhang, C.; Feng, Y.P.; Shen, L. Heterostructures of phosphorene and transition metal dichalcogenides for excitonic solar cells: A first-principles study. Appl. Phys. Lett. 2016, 108, 122105. [Google Scholar] [CrossRef]
- Fei, R.; Faghaninia, A.; Soklaski, R.; Yan, J.-A.; Lo, C.; Yang, L. Enhanced Thermoelectric Efficiency via Orthogonal Electrical and Thermal Conductances in Phosphorene. Nano Lett. 2014, 14, 6393–6399. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Kumar, P.; Thakur, A.; Singh Chauhan, Y.; Bhowmick, S.; Agarwal, A. Anisotropic plasmons, excitons, and electron energy loss spectroscopy of phosphorene. Phys. Rev. B 2017, 96, 035422. [Google Scholar] [CrossRef]
- Jin, F.; Roldán, R.; Katsnelson, M.; Yuan, S. Screening and plasmons in pure and disordered single- and bilayer black phosphorus. Phys. Rev. B 2015, 92, 115440. [Google Scholar] [CrossRef] [Green Version]
- Low, T.; Roldán, R.; Wang, H.; Xia, F.; Avouris, P.; Moreno, L.M.; Guinea, F. Plasmons and Screening in Monolayer and Multilayer Black Phosphorus. Phys. Rev. Lett. 2014, 113, 106802. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zeng, X.C. Bilayer Phosphorene: Effect of Stacking Order on Bandgap and Its Potential Applications in Thin-Film Solar Cells. J. Phys. Chem. Lett. 2014, 5, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Jhun, B.; Park, C.-H. Electronic structure of charged bilayer and trilayer phosphorene. Phys. Rev. B 2017, 96, 085412. [Google Scholar] [CrossRef]
- Qiao, J.; Kong, X.; Hu, Z.-X.; Yang, F.; Ji, W.H. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Mullen, J.T.; Kim, K.W. Highly anisotropic electronic transport properties of monolayer and bilayer phosphorene from first principles. Appl. Phys. Lett. 2016, 109, 053108. [Google Scholar] [CrossRef]
- Caklr, D.; Sevik, C.; Peeters, F.M. Significant effect of stacking on the electronic and optical properties of few-layer black phosphorus. Phys. Rev. B 2015, 92, 165406. [Google Scholar] [CrossRef]
- Torbatian, Z.; Asgari, R. Collective modes in few layer phosphorous. To be submitted 2018, In preparation.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Structures | Lattice | a (Å) | b (Å) | Gap (eV) | MP | ||
|---|---|---|---|---|---|---|---|
| Monolayer | Bilayer | Monolayer | Bilayer | ||||
| graphene | Hexagonal | − | |||||
| MoS | Hexagonal | − | |||||
| phosphorene | Rectangular | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torbatian, Z.; Asgari, R. Plasmonic Physics of 2D Crystalline Materials. Appl. Sci. 2018, 8, 238. https://doi.org/10.3390/app8020238
Torbatian Z, Asgari R. Plasmonic Physics of 2D Crystalline Materials. Applied Sciences. 2018; 8(2):238. https://doi.org/10.3390/app8020238
Chicago/Turabian StyleTorbatian, Zahra, and Reza Asgari. 2018. "Plasmonic Physics of 2D Crystalline Materials" Applied Sciences 8, no. 2: 238. https://doi.org/10.3390/app8020238
APA StyleTorbatian, Z., & Asgari, R. (2018). Plasmonic Physics of 2D Crystalline Materials. Applied Sciences, 8(2), 238. https://doi.org/10.3390/app8020238