Trouton’s Rule for Vapor Sorption in Solids


Abstract
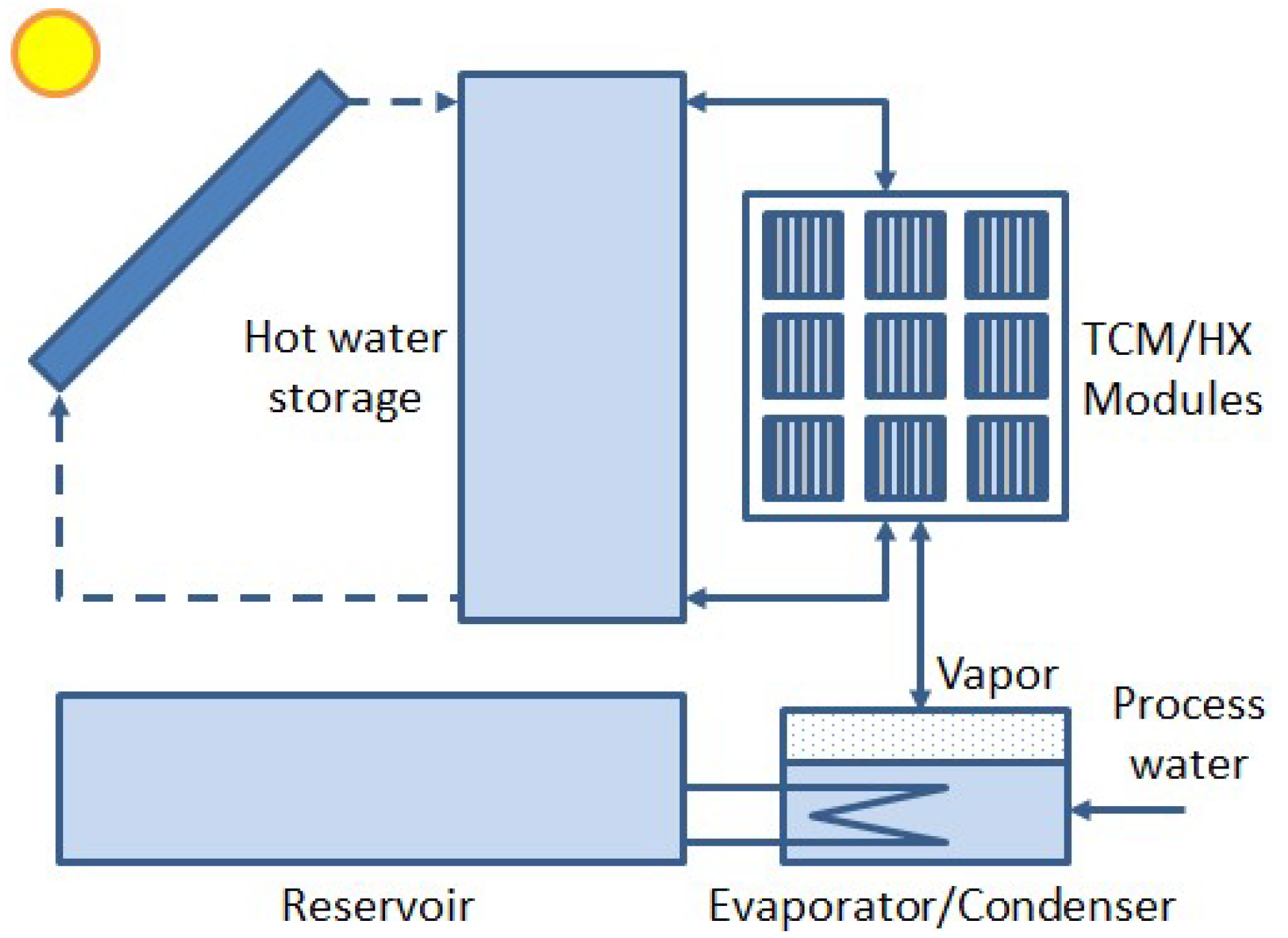
:Featured Application
Abstract

1. Introduction
- Validation of experimentally obtained pT-lines;
- Estimation of pT-lines when only Δh0 is known;
- Predicting limits for the heat storage density of a given desorption temperature.
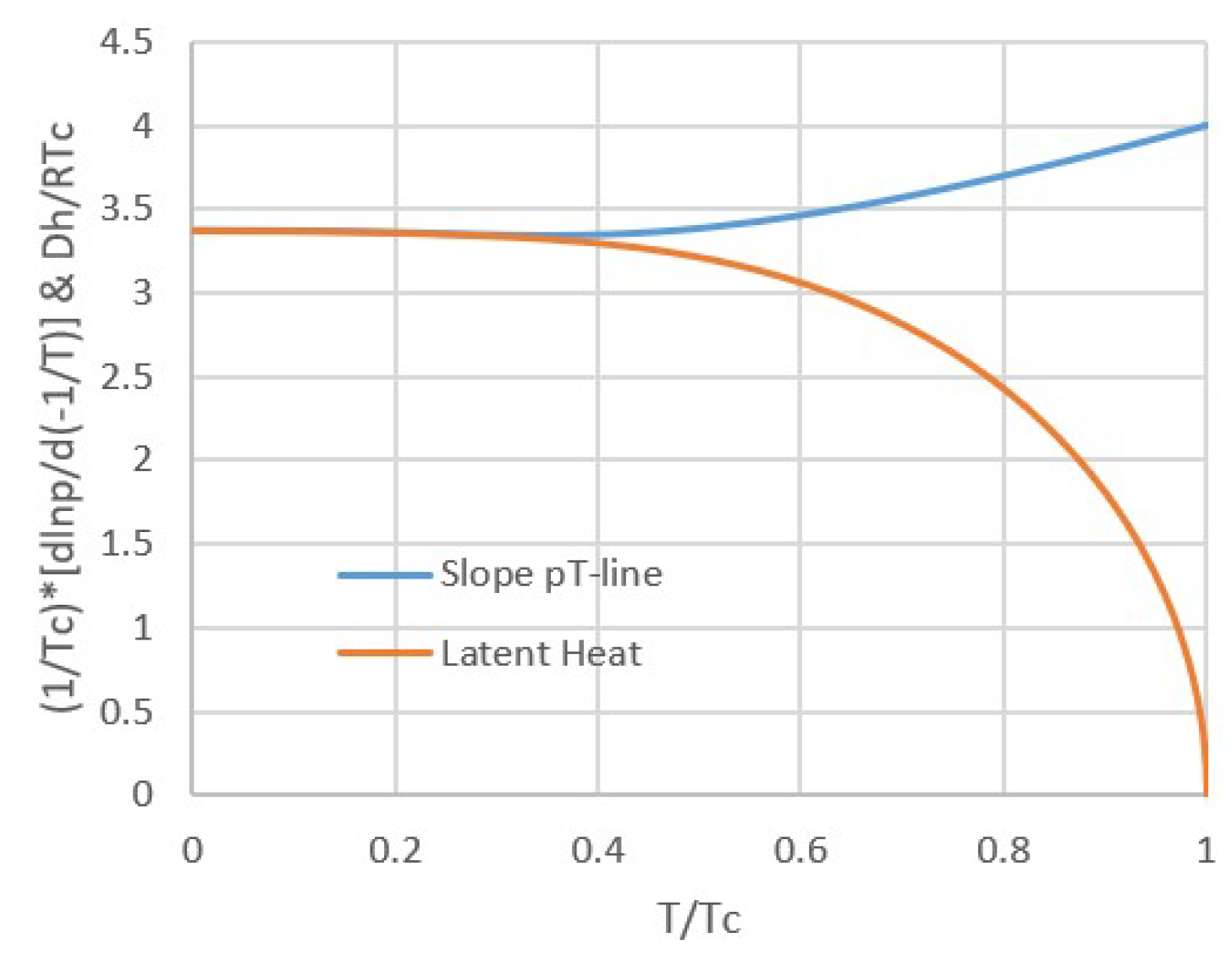
2. Thermodynamic Evidence for Trouton’s Rule
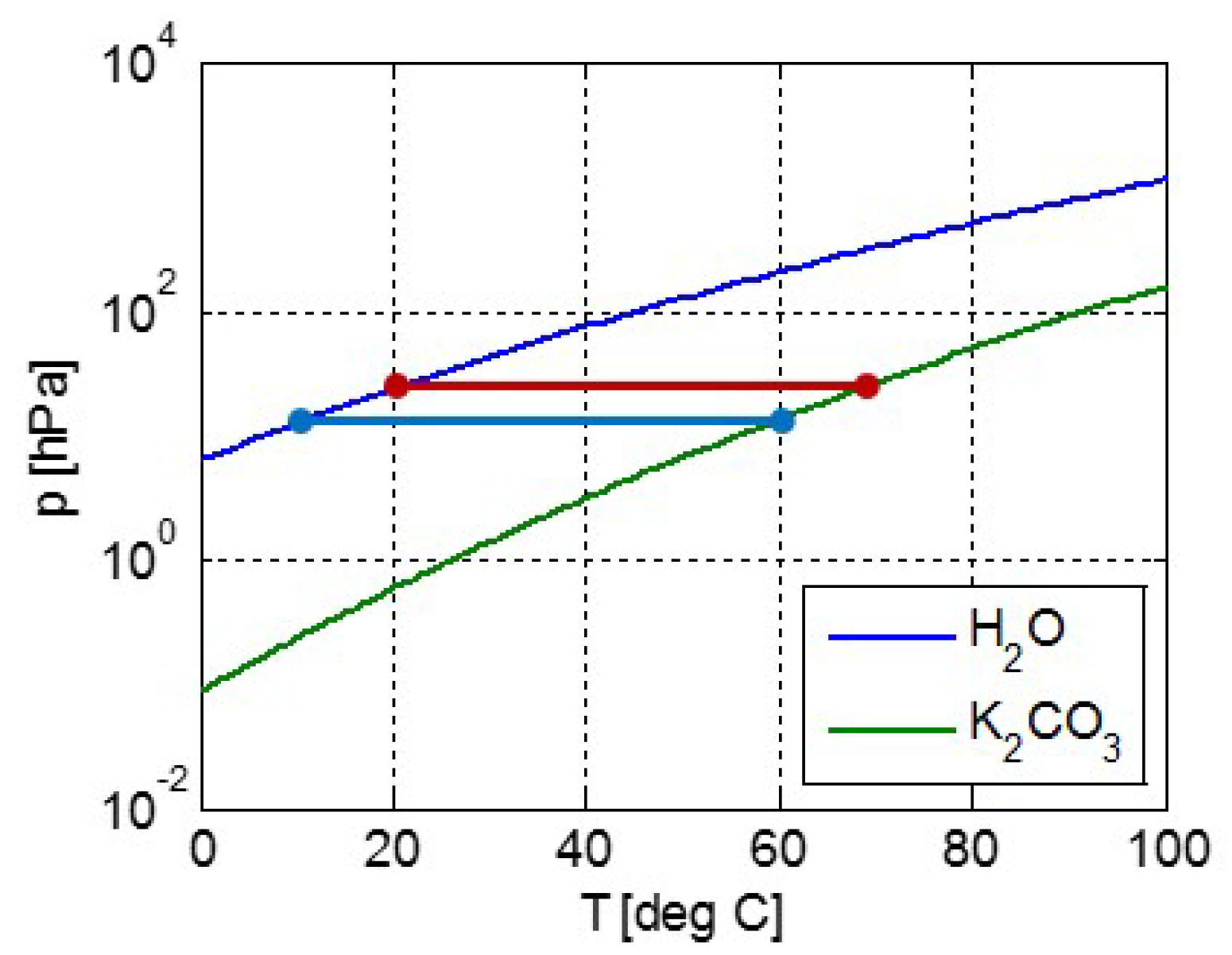
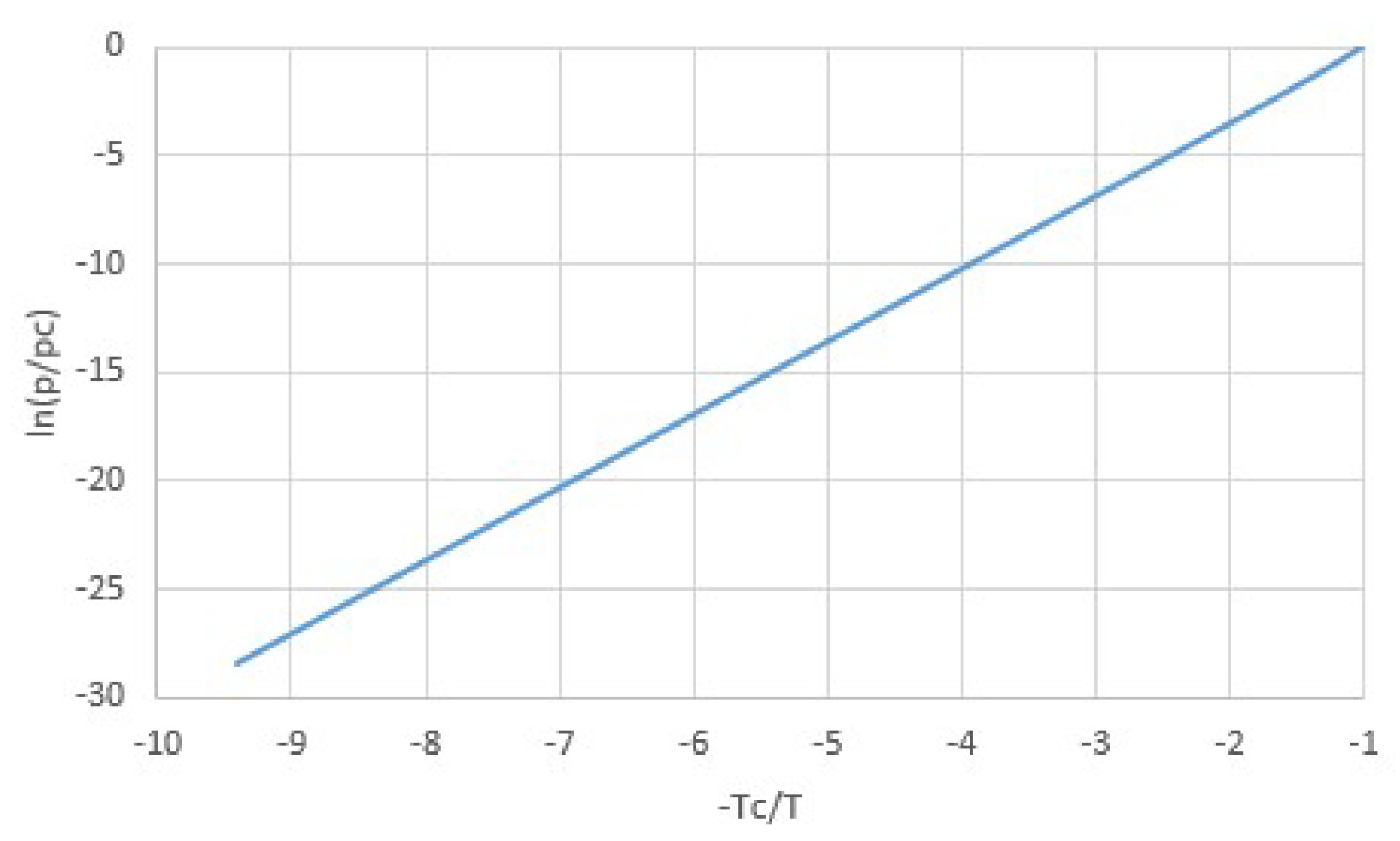
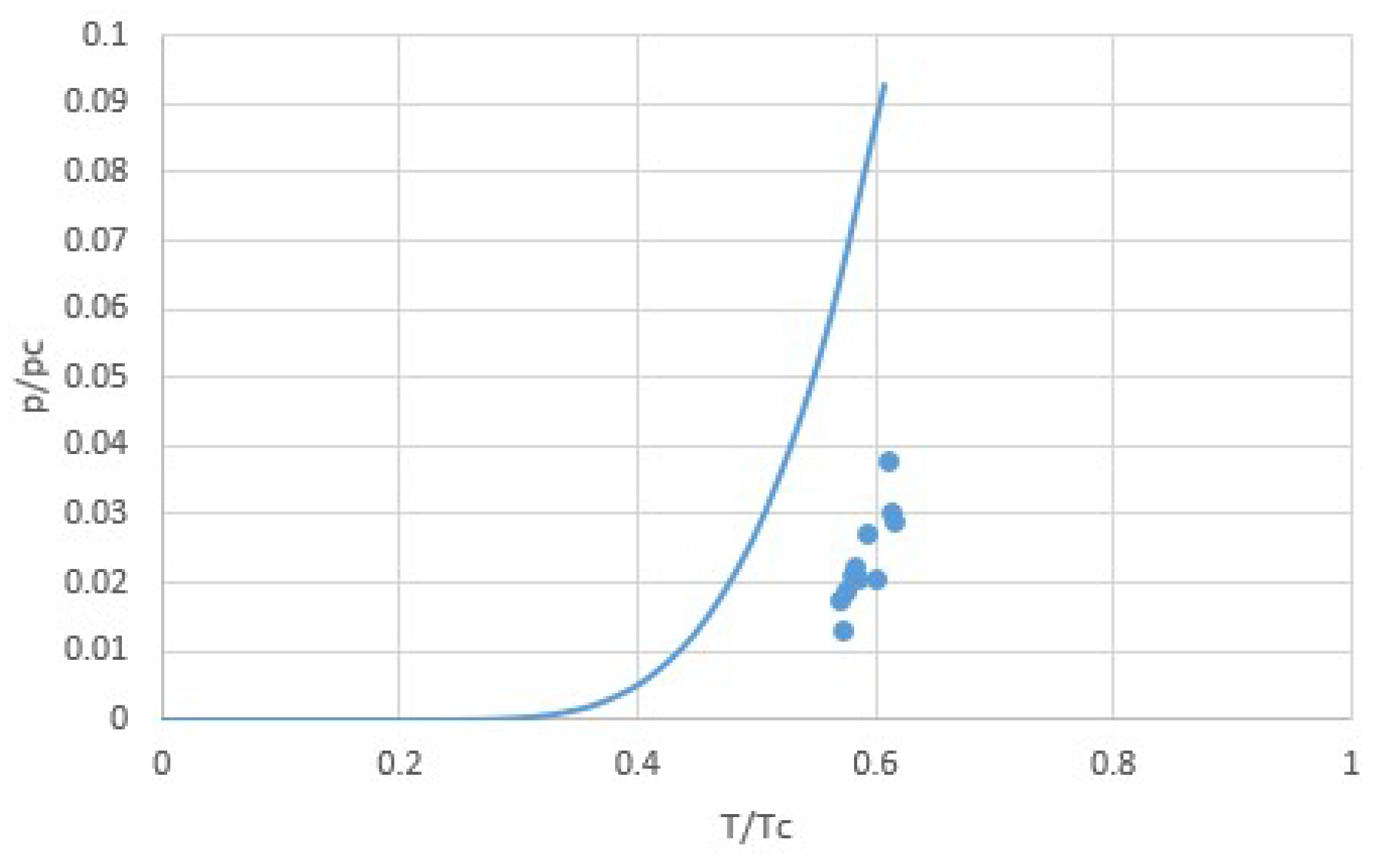
2.1. Modeling pT-Lines
2.2. Trouton’s Rule for Vapor-Liquid and Vapor-Sorber Equilibria
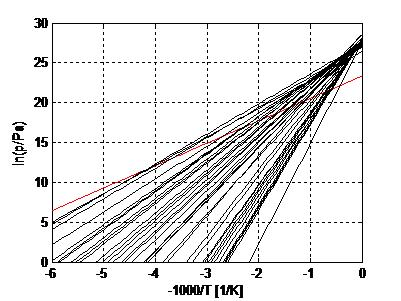
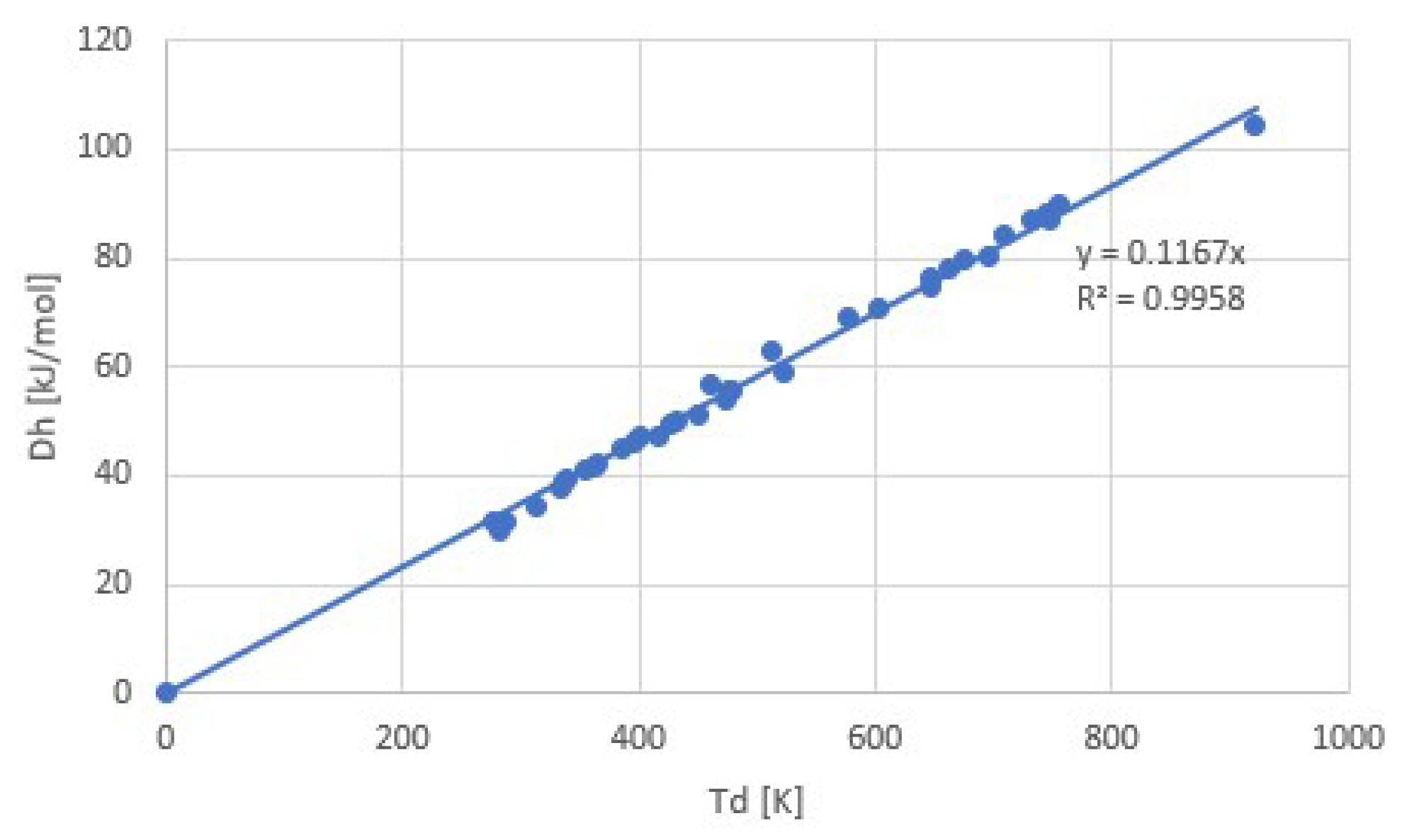
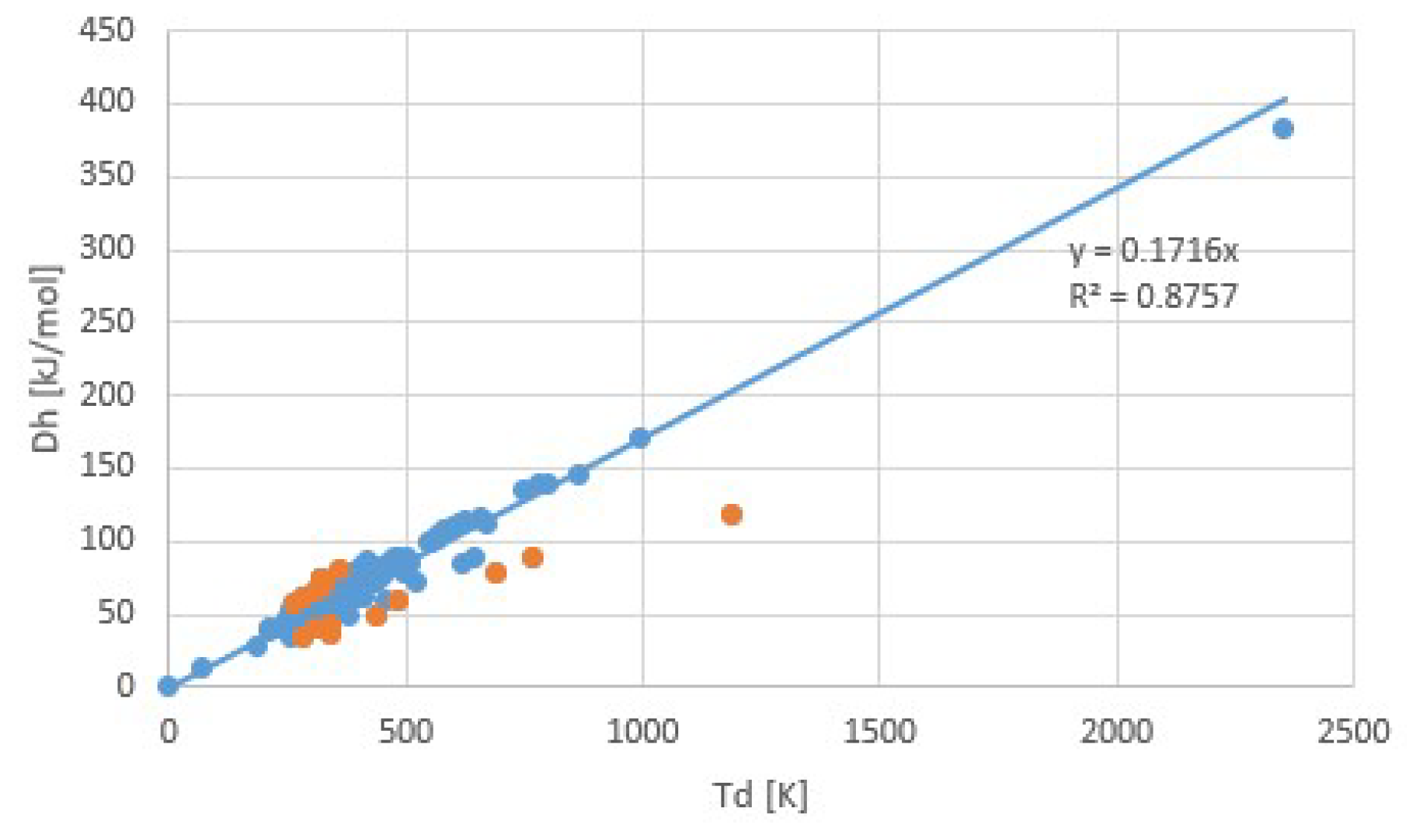
3. Experimental Evidence for Trouton’s Rule
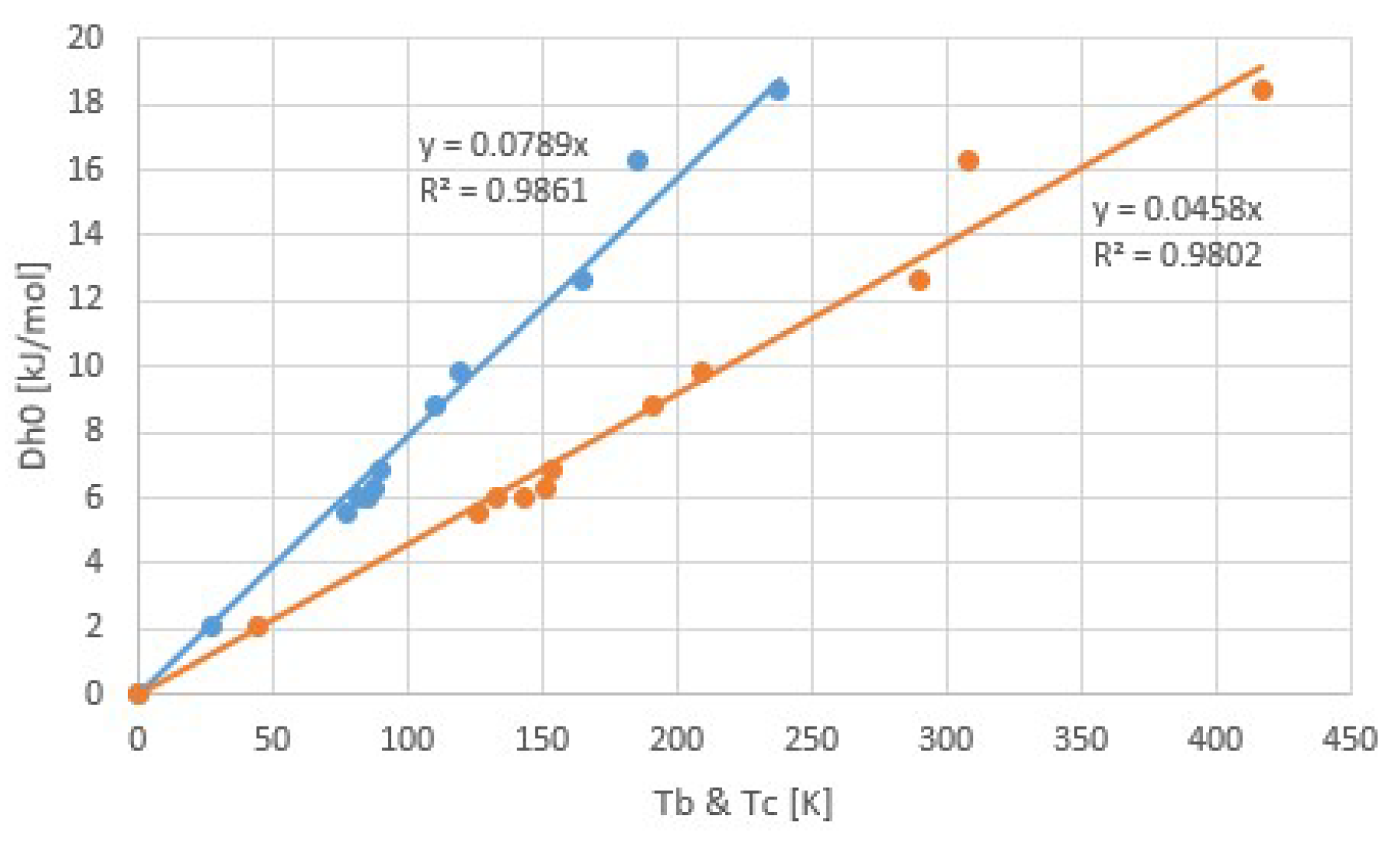
3.1. Common Fluids
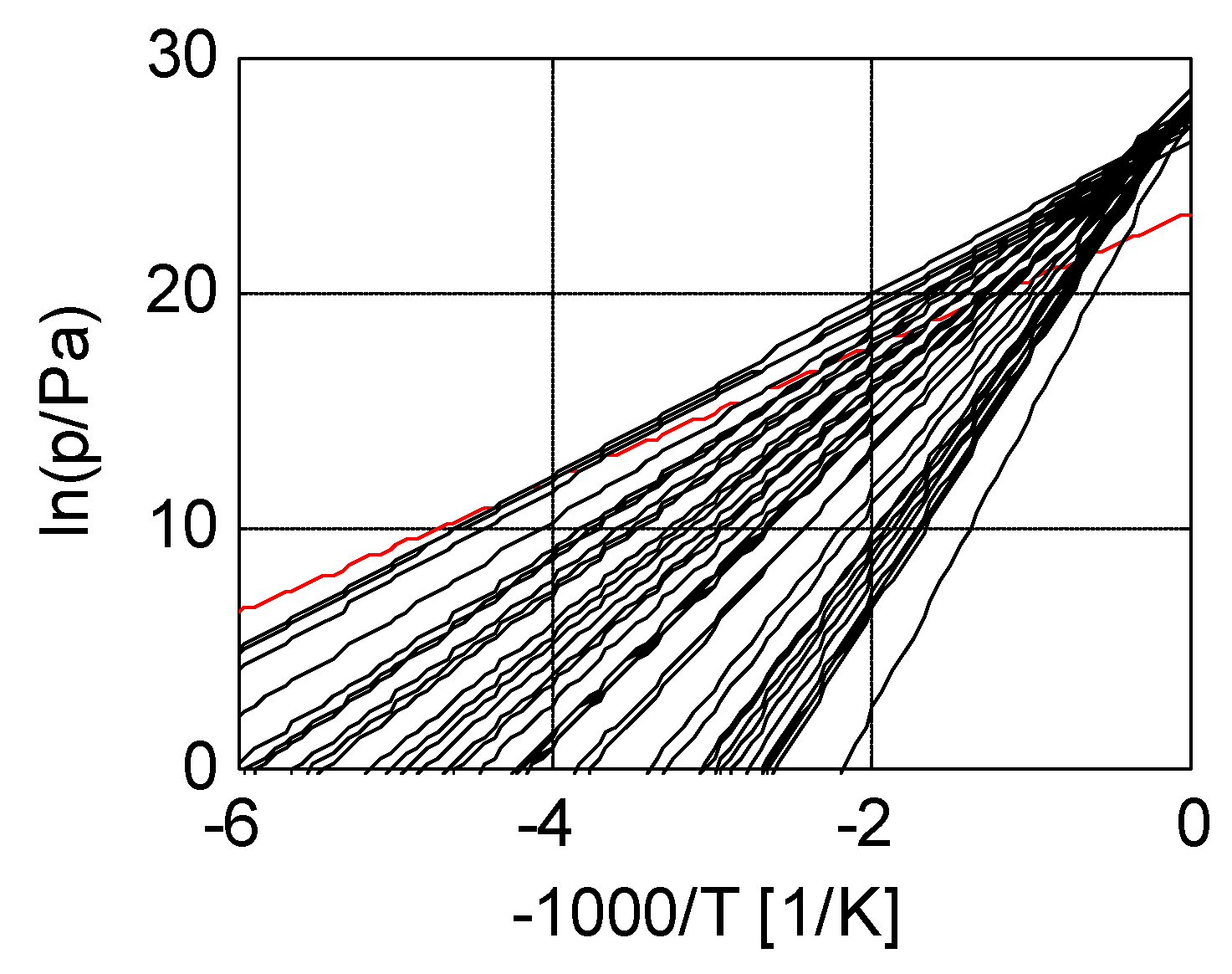
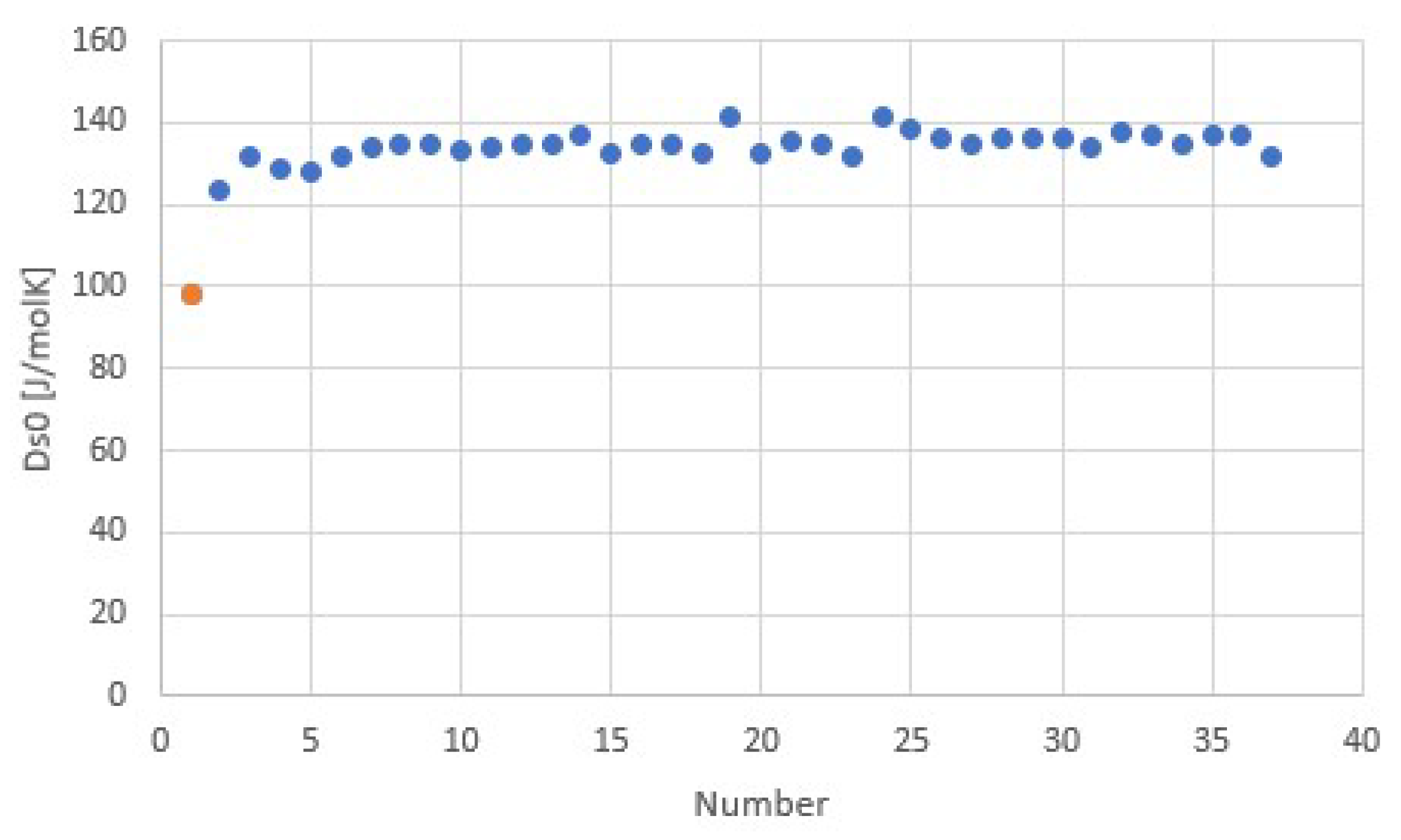
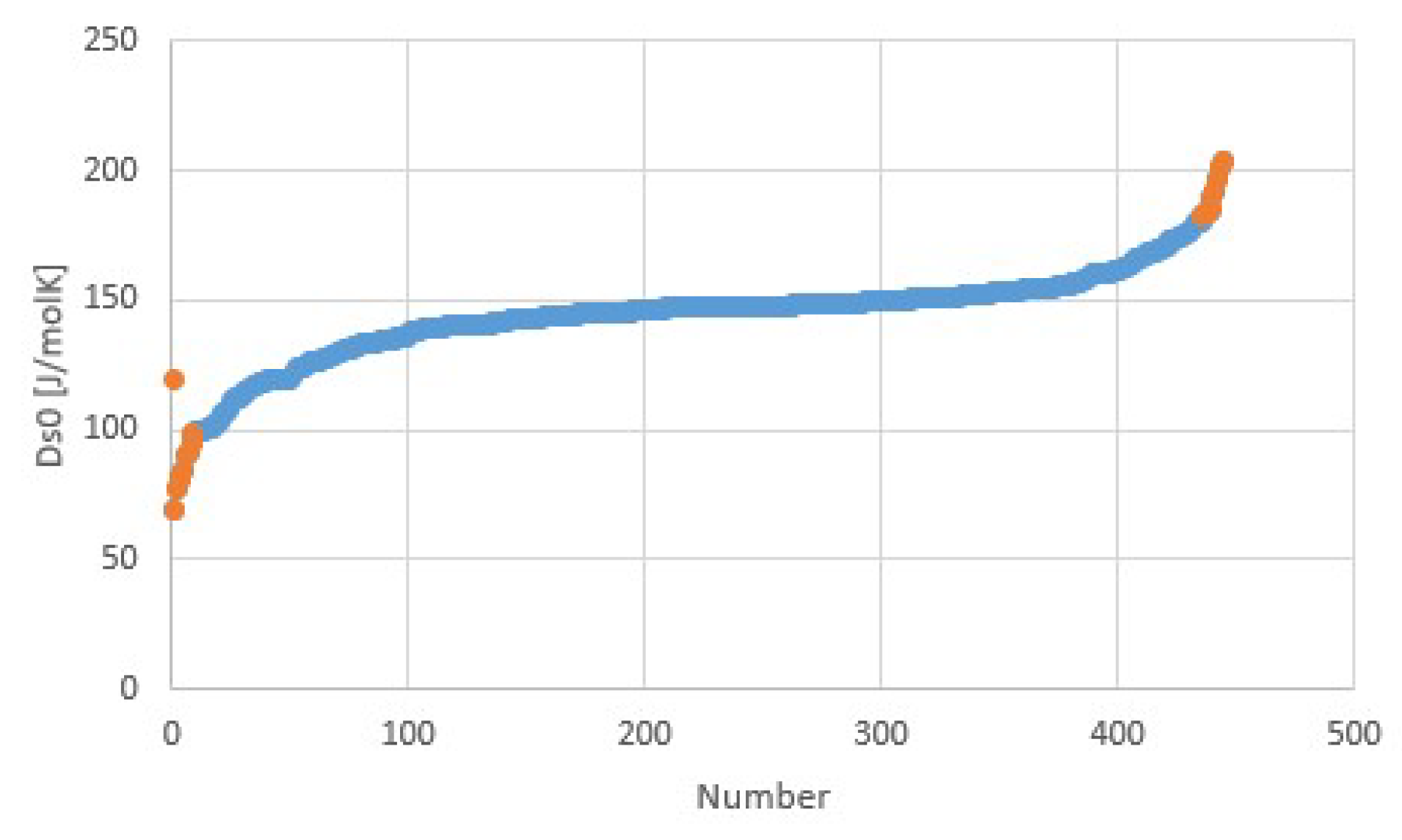
3.2. Salt Ammoniates
3.3. Salt Hydrates
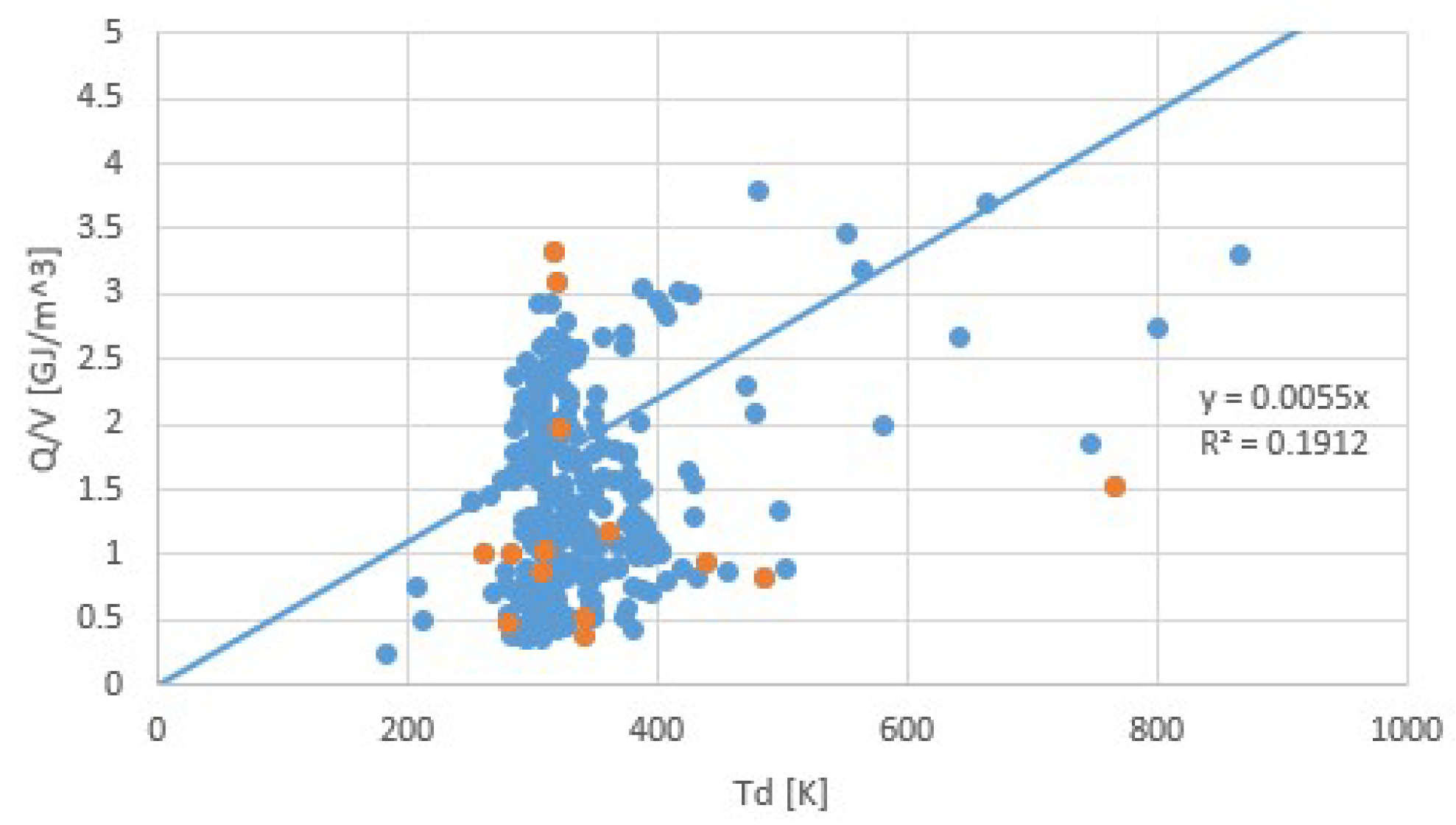
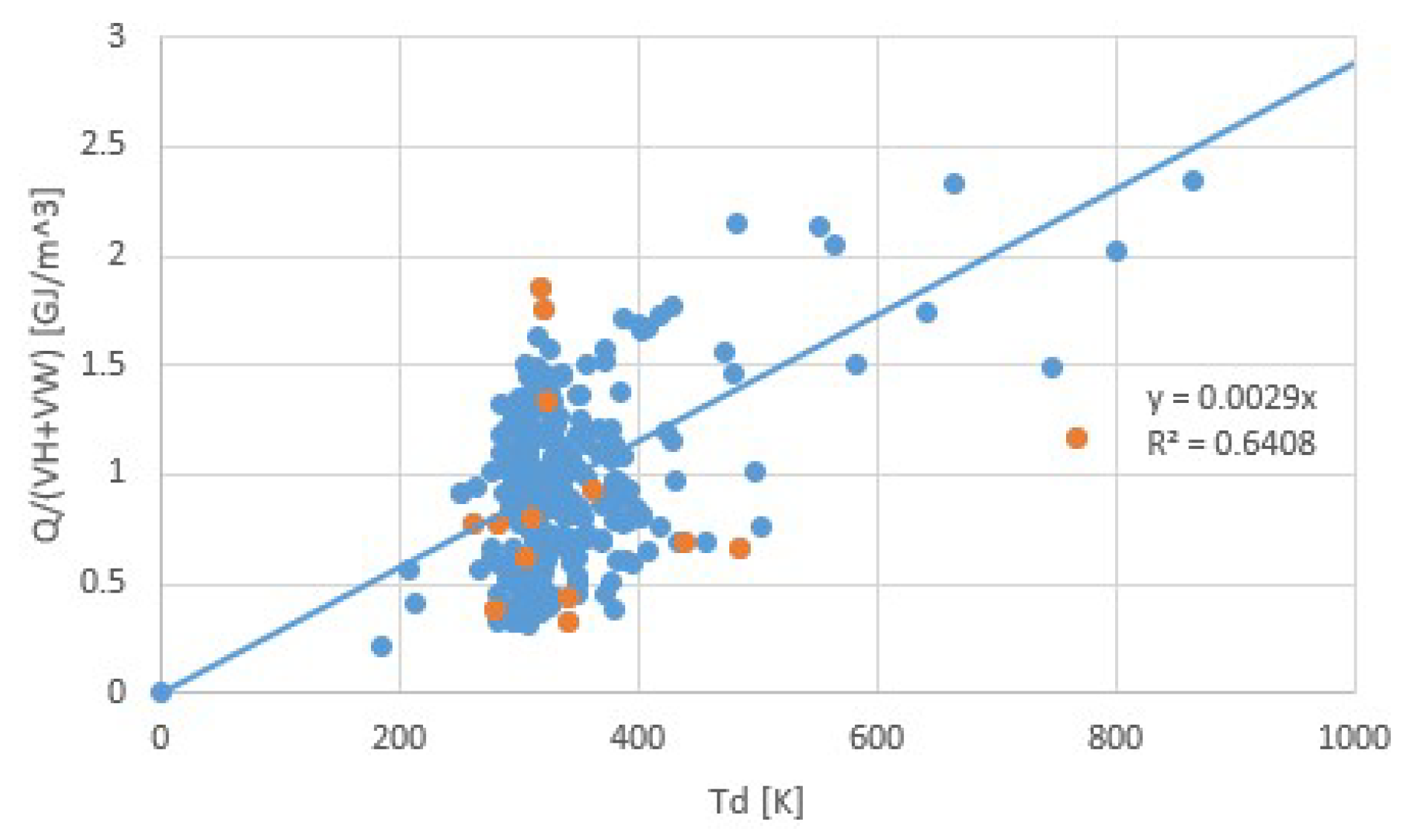
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brunberg, E.A. The TEPIDUS System for Seasonal Heat Storage and for Cooling. In Proceedings of the International Seminar on Thermochemical Heat Storage, Stockholm, Sweden, 7–9 January 1980; Wettermark, G.. pp. 247–260. [Google Scholar]
- Andersson, J.Y.; de Pablo, J.; Azoulay, M. Kinetics of the rehydration of sodium sulphide dehydrated in situ, under formation of its pentahydrate. Thermochim. Acta 1985, 91, 223–234. [Google Scholar] [CrossRef]
- De Boer, R.; Haije, W.; Veldhuis, J. Determination of structural, thermodynamic and phase properties in the Na2S-H2O system for application in a chemical heat pump. Thermochim. Acta 2003, 395, 3–19. [Google Scholar] [CrossRef]
- De Jong, A.J.; Trausel, F.; Finck, C.; van Vliet, L.; Cuypers, R. Thermochemical heat storage—System design issues. Energy Procedia 2014, 48, 309–319. [Google Scholar] [CrossRef]
- Donkers, P.A.J.; Sögütoglu, L.C.; Huinink, H.P.; Fischer, H.; Adan, O.C.G. A review of salt hydrates for seasonal heat storage in domestic applications. Appl. Energy 2017, 199, 45–68. [Google Scholar] [CrossRef]
- N’Tsoukpoe, K.; Liu, H.; Le Pierrès, N.; Luo, L. A review on long-term sorption solar energy storage. Renew. Sustain. Energy Rev. 2009, 13, 2385–2396. [Google Scholar] [CrossRef]
- Cot-Gores, J.; Castell, A.; Cabeza, L.F. Thermochemical energy storage and conversion: A-state-of-the-art review of the experimental research under practical conditions. Renew. Sustain. Energy Rev. 2012, 16, 5207–5224. [Google Scholar] [CrossRef]
- Aristov, Y.I. Challenging offers of material science for adsorption heat transformation: A review. Appl. Thermal Eng. 2013, 50, 1610–1618. [Google Scholar] [CrossRef]
- Solé, A.; Martorell, I.; Cabeza, L.F. State of the art on gas–solid thermochemical energy storage systems and reactors for building applications. Renew. Sustain. Energy Rev. 2015, 47, 386–398. [Google Scholar] [CrossRef]
- Glasser, L. Thermodynamics of Inorganic Hydration and of Humidity Control, with an Extensive Database of Salt Hydrate Pairs. J. Chem. Eng. Data 2014, 59, 526–530. [Google Scholar] [CrossRef]
- Neveu, P.; Castaing, J. Solid-gas chemical heat pumps: Field of application and performance of the internal heat of reaction recovery process. Heat Recovery Syst. CHP 1993, 13, 233–251. [Google Scholar] [CrossRef]
- Halling, P.J. Salt hydrates for water activity control with biocatalysts in organic media. Biotechnol. Tech. 1992, 6, 271–276. [Google Scholar] [CrossRef]
- The International Center for Diffraction Data (ICDD). Available online: www.icdd.com (accessed on 1 December 2017).
- Trouton, F. On Molecular Latent Heat. Philos. Mag. 1884, 18, 54–57. [Google Scholar] [CrossRef]
- Critoph, R.E. Performance limitations of adsorption cycles for solar cooking. Sol. Energy 1988, 41, 21–31. [Google Scholar] [CrossRef]
- Aristov, Y.I.; Tokarev, M.M.; Sharonov, V.E. Universal relation between the boundary temperatures of a basic cycle of sorption heat machines. Chem. Eng. Sci. 2008, 63, 2907–2912. [Google Scholar] [CrossRef]
- Zemansky, M.W.; Dittman, M. Heat and Thermodynamics, 6th ed.; McGraw-Hill Book Company: Tokyo, Japan, 1981; ISBN-13: 978-0070666474. [Google Scholar]
- Van der Waals, J.D. On the Continuity of the Gaseous and Liquid State. Ph.D. Thesis, University of Leiden, Leiden, The Netherlands, 1873. [Google Scholar]
- Tabor, D. Gases, Liquids and Solids, 2nd ed.; Cambridge University Press: Cambridge, UK, 1979; ISBN-13: 978-0521294669. [Google Scholar]
- Lekner, J. Parametric solution of the van dere Waals liquid-vapor coexistence curve. Am. J. Phys. 1982, 50, 161–163. [Google Scholar] [CrossRef]
- Johnston, D.C. Thermodynamic Properties of the van der Waals fluid. arXiv 2014. [Google Scholar]
- Polanyi, M.; Wigner, E.P. Über die Interferenz von Eigenschwingungen las Ursache von Energieschwankungen und chemischer Umsetzungen. Z. Phys. Chem. A 1928, 43, 439–452. [Google Scholar]
- Kittel, C.; Kroemer, H. Thermal Physics, 2nd ed.; W.H. Freeman & Company: San Francisco, CA, USA, 1980; ISBN-13: 978-0716710882. [Google Scholar]
- Guggenheim, E.A. The Principle of Corresponding States. J. Chem. Phys. 1945, 13, 253–261. [Google Scholar] [CrossRef]
- Hildebrand, J.H. The entropy of vaporization as a means of distinguishing normal liquids. J. Am. Chem. Soc. 1915, 37, 970–978. [Google Scholar] [CrossRef]
- Everett, D.H. Some Correlations between Thermodynamic Porperties and the Structure of Liquids. J. Chem. Soc. 1960, 2566–2573. [Google Scholar] [CrossRef]
- Donkers, P.A.J. Experimental Study on Thermochemical Heat Storage Materials. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2015. [Google Scholar]
- Springer Materials. Available online: http://materials.springer.com/periodictable# (accessed on 15 December 2017).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluid | Tb (K) | Tc (K) | Δh (J/molK) | Δh/RTb | Δh/RTc |
|---|---|---|---|---|---|
| H2O | 373.15 | 647.6 | 44.0 | 14.2 | 8.2 |
| NH3 | 239.75 | 405.5 | 23.35 | 11.7 | 6.9 |
| Salt | H2O | Δs (J/molK) | Alternative Δs (J/molK) |
|---|---|---|---|
| Ca(H2PO4)2 | 1-0 | 69 | |
| MnI2 | 2-1 | 77 | |
| CuSO4 | 1-0 | 81 | |
| MnSeO4 | 1-0 | 82 | |
| MgSeO4 | 1-0 | 84 | |
| CsCd(SO4)2 | 6-2 | 90 | |
| SrCl2 | 2-1 | 91 | 148 |
| MnI2 | 1-0 | 95 | |
| Na2HPO4 | 12-7 | 99 | 149 |
| CaCl2 | 6-4 | 182 | 142, 166 |
| SrCl2 | 1-0 | 184 | |
| Tl(Al(SO4))2 | 12-0 | 184 | |
| LiBr | 2-1 | 185 | 171 |
| BaCl2 | 1-0 | 190 | 100, 145 |
| Na2B4O7 | 5-0 | 192 | 149 for 10-0 |
| CsCr(SeO4)2 | 12-0 | 196 | |
| HIO3 | 1-0 | 201 | |
| CH3NH2Cr(SO4)2 | 12-0 | 204 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Jong, A.-J.; Fischer, H. Trouton’s Rule for Vapor Sorption in Solids. Appl. Sci. 2018, 8, 638. https://doi.org/10.3390/app8040638
De Jong A-J, Fischer H. Trouton’s Rule for Vapor Sorption in Solids. Applied Sciences. 2018; 8(4):638. https://doi.org/10.3390/app8040638
Chicago/Turabian StyleDe Jong, Ard-Jan, and Hartmut Fischer. 2018. "Trouton’s Rule for Vapor Sorption in Solids" Applied Sciences 8, no. 4: 638. https://doi.org/10.3390/app8040638
APA StyleDe Jong, A. -J., & Fischer, H. (2018). Trouton’s Rule for Vapor Sorption in Solids. Applied Sciences, 8(4), 638. https://doi.org/10.3390/app8040638