Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review




Abstract
:Featured Application
Abstract

1. Introduction
2. Theoretical Studies on Heteroatom-Mediated Metal–Support Interactions
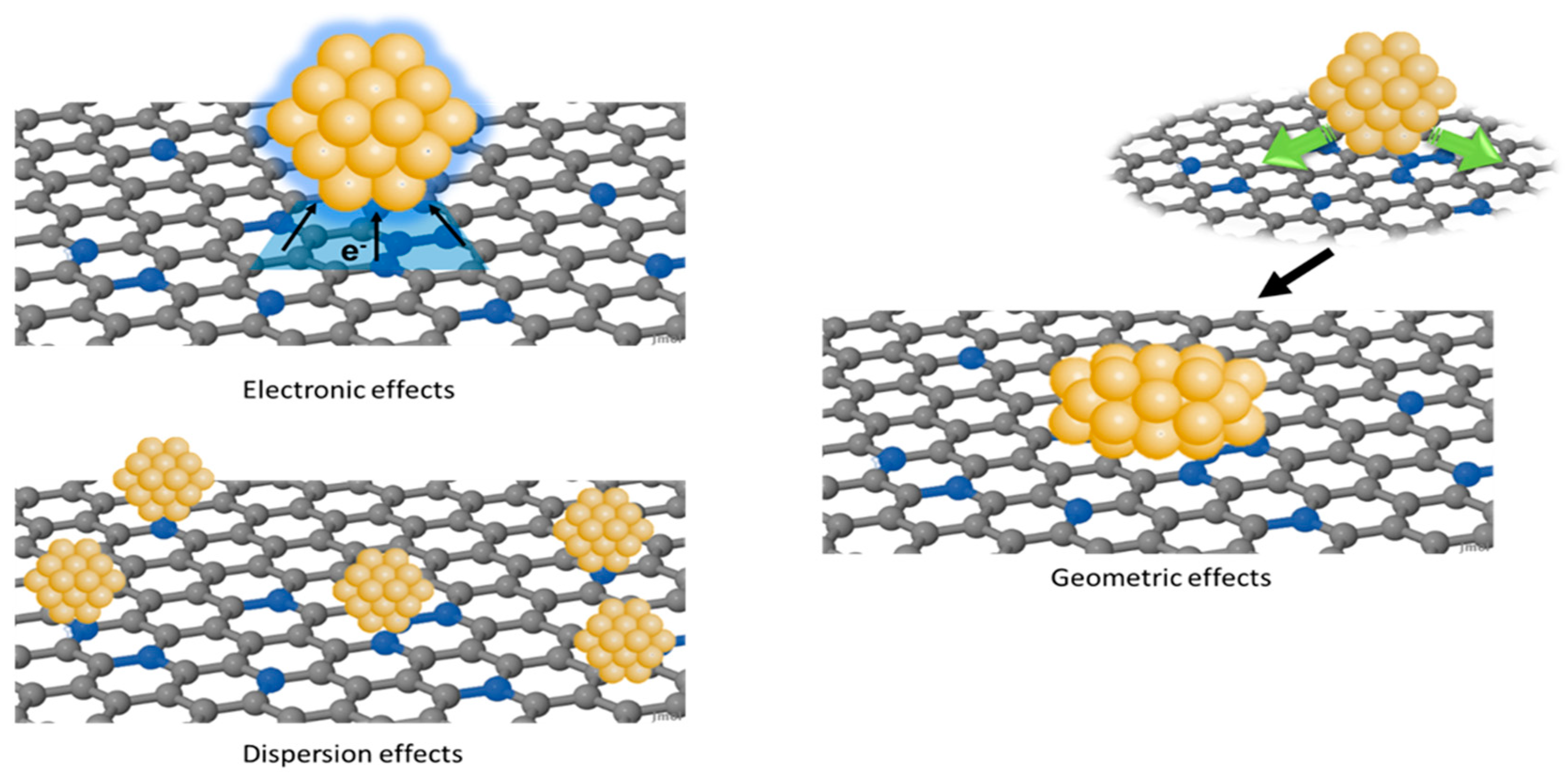
2.1. Basic Concepts for Understanding Heteroatom Mediated Metal–Support Interactions
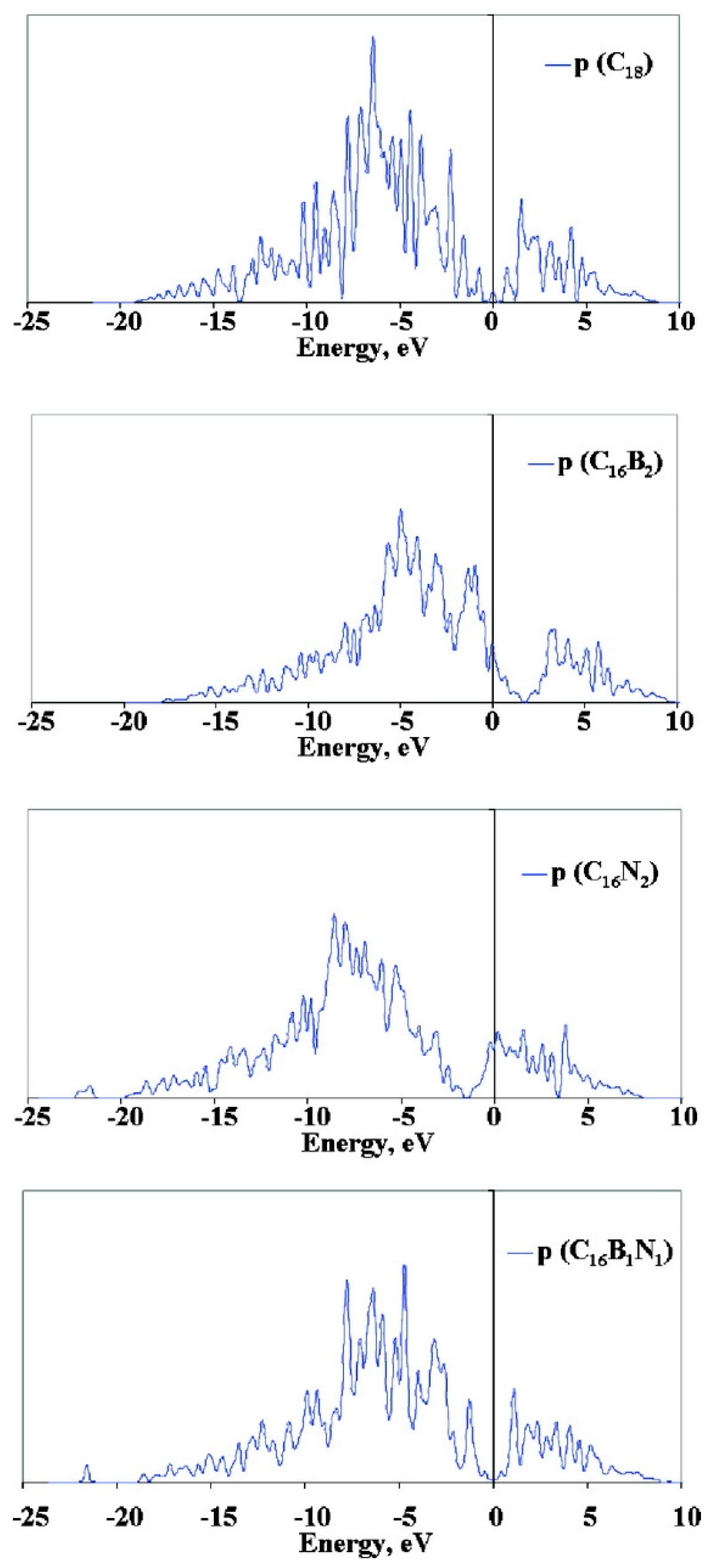
2.2. Metal Single-Atom Adsorption on Doped Carbon Surfaces
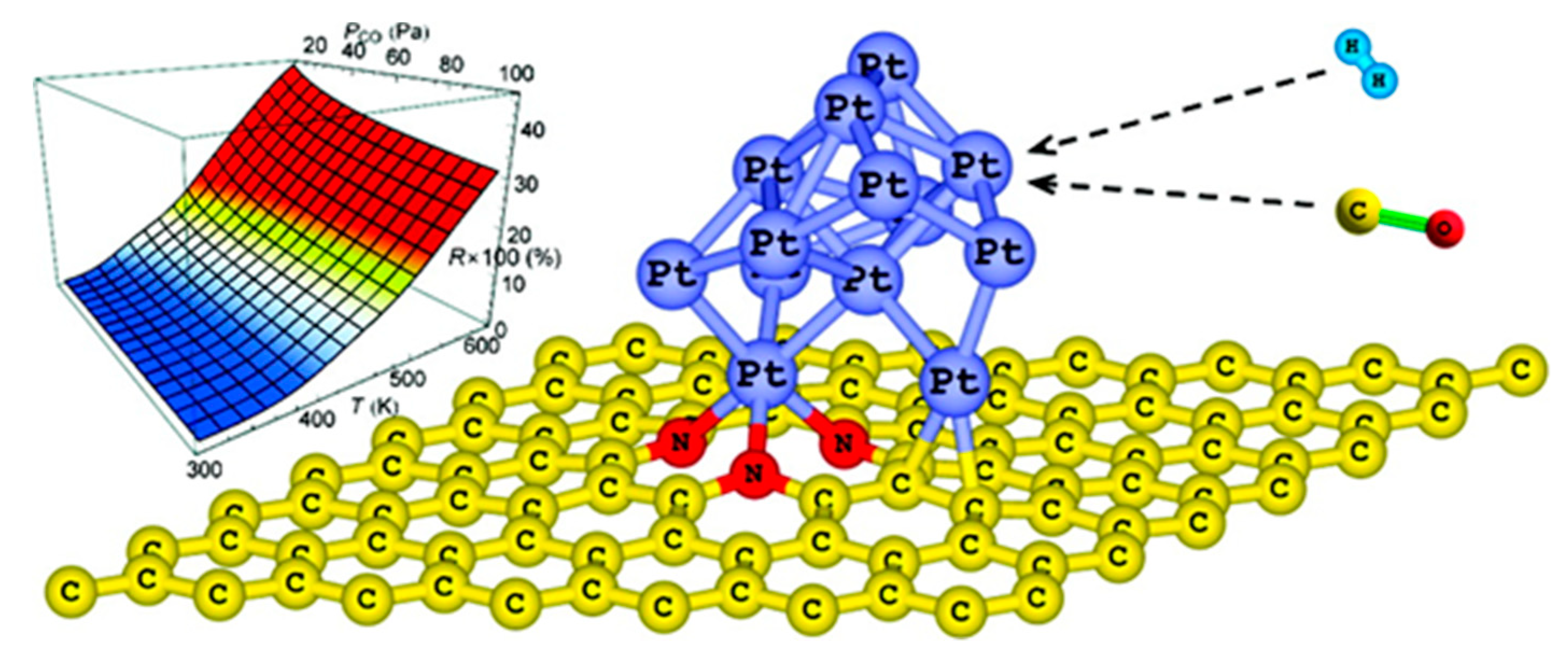
2.3. From Single Metal Atoms to Metal Clusters
- (a)
- the match between the electronic structure of the metal and that of the support
- (b)
- the number of bonds that are created with the surface atoms (carbon and heteroatoms)
- (c)
- breaking of the long-range electronic delocalization
- (d)
- local surface deformation, corrugation, and defect introduction
- (e)
- electron density redistribution and charge transfer
- (f)
- the presence of atoms near the adsorption sites that are available to saturate the eventually dangling bonds.
2.4. Predicting the Effect of Heteroatom-Mediated SMSI on the Catalytic Performances
2.4.1. Effects on Molecule Adsorption and Metal Cluster Shape
2.4.2. SMSI and Metal Dispersion
3. Probing the Metal–Support Interactions at the Interface
3.1. X-ray Photoelectron Spectroscopy (XPS)
3.2. X-ray Absorption Spectroscopy (XAS)
3.3. Transmission Electron Microscopies and Other Imaging Techniques
3.4. Other Techniques: Electrochemical and Adsorption Properties Characterization
4. Effect of Heteroatoms–Metal Interaction in the Liquid Phase Oxidation of Benzyl Alcohol
5. Conclusions
Funding
Conflicts of Interest
References
- Ebert, L.B. Science of fullerenes and carbon nanotubes. Carbon 1997, 35, 437–438. [Google Scholar] [CrossRef]
- Hirsch, A. The chemistry of fullerenes: An overview. Angew. Chem. Int. Ed. 1993, 32, 1138–1141. [Google Scholar] [CrossRef]
- Guldi, D.M.; Martìn, N. Carbon Nanotubes and Related Structures: Production and Formation; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- De Jong, K.P.; Geus, J.W.; Jong, K. De Carbon Nanofibers: Catalytic Synthesis and Applications. Catal. Rev. 2000, 42, 481–510. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and Prospects. Prospects 2009, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yan, J.; Fan, Z. Carbon materials for high volumetric performance supercapacitors: Design, progress, challenges and opportunities. Energy Environ. Sci. 2016, 9, 729–762. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Yan, M.; Yu, J. Carbon nanostructures in biology and medicine. J. Mater. Chem. B 2017, 5, 6437–6450. [Google Scholar] [CrossRef]
- Zieleniewska, A.; Lodermeyer, F.; Roth, A.; Guldi, D.M. Fullerenes-how 25 years of charge transfer chemistry have shaped our understanding of (interfacial) interactions. Chem. Soc. Rev. 2018, 47, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.-O.; Ralph, S.F. Carbon Nanotube Membranes: Synthesis, Properties, and Future Filtration Applications. Nanomaterials 2017, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Dou, Y.; Zhao, D.; Fulvio, P.F.; Mayes, R.T.; Dai, S. Carbon materials for chemical capacitive energy storage. Adv. Mater. 2011, 23, 4828–4850. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Z.; Chen, Y.M.; Xia, Y.; Liang, W.; Zhu, Y.; Vogt, B.D. Ultra-long cycle life, low-cost room temperature sodium-sulfur batteries enabled by highly doped (N, S) nanoporous carbons. Nano Energy 2017, 32, 59–66. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liang, W.; Li, S.; Zou, F.; Bhaway, S.M.; Qiang, Z.; Gao, M.; Vogt, B.D.; Zhu, Y. A nitrogen doped carbonized metal-organic framework for high stability room temperature sodium-sulfur batteries. J. Mater. Chem. A 2016, 4, 12471–12478. [Google Scholar] [CrossRef]
- Liu, X.; Zou, F.; Liu, K.; Qiang, Z.; Taubert, C.J.; Ustriyana, P.; Vogt, B.D.; Zhu, Y. A binary metal organic framework derived hierarchical hollow Ni3S2/Co9S8/N-doped carbon composite with superior sodium storage performance. J. Mater. Chem. A 2017, 5, 11781–11787. [Google Scholar] [CrossRef]
- Serp, B.; Machado, B. Nanostructured Carbon Materials for Catalysis; RSC Catalysis Series No. 23; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Chemical Functionalization of Carbon Nanomaterials Chemistry and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Hirsch, A. Functionalization of single-walled carbon nanotubes. Angew. Chem. Int. Ed. 2002, 41, 1853–1859. [Google Scholar] [CrossRef]
- Wei, D.; Liu, Y.; Wang, Y.; Zhang, H.; Huang, L.; Yu, G. Synthesis of N-Doped Graphene by Chemical Vapor Deposition and Its Electrical Properties. Nano Lett. 2009, 9, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Duclaux, L. Review of the doping of carbon nanotubes (multiwalled and single-walled). Carbon 2002, 40, 1751–1764. [Google Scholar] [CrossRef]
- Park, Y.; Yoo, J.; Lim, B.; Kwon, W.; Rhee, S.-W. Improving the functionality of carbon nanodots: Doping and surface functionalization. J. Mater. Chem. A 2016, 4, 11582–11603. [Google Scholar] [CrossRef]
- Ewels, C.P.; Glerup, M. Nitrogen Doping in Carbon Nanotubes. J. Nanosci. Nanotechnol. 2005, 5, 1345–1363. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kahn, M.G.C.; Wong, S.S. Rational chemical strategies for carbon nanotube functionalization. Chemistry 2003, 9, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.V.; Cividanes, L.D.S.; Brito, F.S.; Menezes, B.R.C.D.; Franceschi, W.; Simonetti, E.A.N.; Thim, G.P. Functionalizing Graphene and Carbon Nanotubes: A Review; Springer: New York, NY, USA, 2016. [Google Scholar]
- Georgakilas, V.; Tiwari, J.N.; Kemp, K.C.; Perman, J.A.; Bourlinos, A.B.; Kim, K.S.; Zboril, R. Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications. Chem. Rev. 2016, 116, 5464–5519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Stoddart, J.F. Noncovalent functionalization of single-walled carbon nanotubes. Acc. Chem. Res. 2009, 42, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Dyke, C.A.; Tour, J.M. Covalent functionalization of single-walled carbon nanotubes for materials applications. J. Phys. Chem. A 2004, 108, 11151–11159. [Google Scholar] [CrossRef]
- Georgakilas, V.; Kordatos, K.; Prato, M.; Guldi, D.M.; Holzinger, M.; Hirsch, A. Organic functionalization of carbon nanotubes. J. Am. Chem. Soc. 2002, 124, 760–761. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Alemany, L.B.; Margrave, J.L.; Khabashesku, V.N. Sidewall Carboxylic Acid Functionalization of Single-Walled Carbon Nanotubes Sidewall Carboxylic Acid Functionalization of Single-Walled Carbon Nanotubes. J. Am. Chem. Soc. 2003, 15174–15182. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Wang, Z.; Fierke, M.A. Functionalization of porous carbon materials with designed pore architecture. Adv. Mater. 2009, 21, 265–293. [Google Scholar] [CrossRef]
- Campisi, S.; Marzorati, S.; Spontoni, P.; Chan-Thaw, C.E.; Longhi, M.; Villa, A.; Prati, L. Tailored N-Containing Carbons as Catalyst Supports in Alcohol Oxidation. Materials 2016, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Nxumalo, E.N.; Coville, N.J. Nitrogen doped carbon nanotubes from organometallic compounds: A review. Materials 2010, 3, 2141–2171. [Google Scholar] [CrossRef]
- Prati, L.; Chan-Thaw, C.E.; Campisi, S.; Villa, A. N-Modified Carbon-Based Materials: Nanoscience for Catalysis. Chem. Rec. 2016, 16, 2187–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, S.; Morin, S.; Stevenson, K.J. Structure, composition, and chemical reactivity of carbon nanotubes by selective nitrogen doping. Carbon 2006, 44, 1429–1437. [Google Scholar] [CrossRef]
- Strano, M.S.; Dyke, C.A.; Usrey, M.L.; Barone, P.W.; Allen, M.J.; Shan, H.; Kittrell, C.; Hauge, R.H.; Tour, J.M.; Smalley, R.E. Electronic structure control of single-walled carbon nanotube functionalization. Science 2003, 301, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Santos, C.; Martínez-Hernández, A.L.; Fisher, F.T.; Ruoff, R.S.; Castaño, V.M. Improvement of Thermal and Mechanical Properties of Carbon Nanotube Composites through Chemical Functionalization. Chem. Mater. 2003, 15, 4470–4475. [Google Scholar] [CrossRef]
- Qiang, Z.; Xia, Y.; Xia, X.; Vogt, B.D. Generalized synthesis of a family of highly heteroatom-doped ordered mesoporous carbons. Chem. Mater. 2017, 29, 10178–10186. [Google Scholar] [CrossRef]
- Lee, W.J.; Maiti, U.N.; Lee, J.M.; Lim, J.; Han, T.H.; Kim, S.O. Nitrogen-doped carbon nanotubes and graphene composite structures for energy and catalytic applications. Chem. Commun. 2014, 50, 6818–6830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, H.; Lu, Y.; Wei, S.; Wujcik, E.; Guo, Z. Multifunctional Carbon Nanostructures for Advanced Energy Storage Applications. Nanomaterials 2015, 5, 755–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Tong, X.; Zhang, G.; Qiao, J.; Gong, Q.; Sun, S. Nitrogen-Doped Carbon Nanotube and Graphene Materials for Oxygen Reduction Reactions. Catalysts 2015, 5, 1574–1602. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mayoral, E.; Calvino-Casilda, V.; Soriano, E. Metal-supported carbon-based materials: Opportunities and challenges in the synthesis of valuable products. Catal. Sci. Technol. 2016, 6, 1265–1291. [Google Scholar] [CrossRef]
- Tauster, S.J. Strong metal-support interactions. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Nicole, J.; Comninellis, C.; Tsiplakides, D.; Pliangos, C.; Verykios, X.E.; Vayenas, C.G. Electrochemical promotion and metal-support interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Ryndin, Y.A.; Hicks, R.F.; Bell, A.T.; Yermakov, Y.I. Effects of metal-support interactions on the synthesis of methanol over palladium. J. Catal. 1981, 70, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Román-Martínez, M.C.; Cazorla-Amorós, D.; Linares-Solano, A.; De Lecea, C.S.M.; Yamashita, H.; Anpo, M. Metal-support interaction in Pt/C catalysts. Influence of the support surface chemistry and the metal precursor. Carbon 1995, 33, 3–13. [Google Scholar] [CrossRef]
- Fujiwara, K.; Okuyama, K.; Pratsinis, S.E. Metal-support interactions in catalysts for environmental remediation. Environ. Sci. Nano 2017, 4, 2076–2092. [Google Scholar] [CrossRef]
- Joyner, W.; Pendry, J.B.; Tennison, R.; Joyner, R.W.; Pendry, J.B.; Saldin, D.K.; Tennison, S.R. Metal-support interactions in heterogeneous catalysis. Surf. Sci. 1984, 138, 84–94. [Google Scholar] [CrossRef]
- Akubuiro, E.C.; Verykios, X.E. Effects of dopants on performance of metal crystallites. Further characterization of doped supports and catalysts. J. Catal. 1988, 113, 106–119. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Zhang, S.; DeRita, L.; Marinkovic, N.S.; Chen, J.G.; Graham, G.W.; Pan, X.; Christopher, P. Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 2017, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Xia, W. Interactions between metal species and nitrogen-functionalized carbon nanotubes. Catal. Sci. Technol. 2016, 6, 630–644. [Google Scholar] [CrossRef]
- Hu, P.; Huang, Z.; Amghouz, Z.; Makkee, M.; Xu, F.; Kapteijn, F.; Dikhtiarenko, A.; Chen, Y.; Gu, X.; Tang, X. Electronic metal-support interactions in single-atom catalysts. Angew. Chem. Int. Ed. 2014, 53, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Vayenas, C.G.; Bebelis, S.; Pliangos, C.; Brosda, S.; Tsiplakides, D. Electrochemical Activation of Catalysis: Promotion, Electrochemical Promotion, and Metal-Support Interactions; Springer Science & Business Media: New York, NY, USA, 2001. [Google Scholar]
- Arrigo, R.; Wrabetz, S.; Schuster, M.E.; Wang, D.; Villa, A.; Rosenthal, D.; Girsgdies, F.; Weinberg, G.; Prati, L.; Schlögl, R.; et al. Tailoring the morphology of Pd nanoparticles on CNTs by nitrogen and oxygen functionalization. Phys. Chem. Chem. Phys. 2012, 14, 10523–10532. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Eitan, A.; Schadler, L.S.; Ajayan, P.M.; Siegel, R.W.; Grobert, N.; Mayne, M.; Reyes-Reyes, M.; Terrones, H.; Terrones, M. Selective attachment of gold nanoparticles to nitrogen-doped carbon nanotubes. Nano Lett. 2003, 3, 275–277. [Google Scholar] [CrossRef]
- Lepró, X.; Terrés, E.; Vega-Cantú, Y.; Rodríguez-Macías, F.J.; Muramatsu, H.; Kim, Y.A.; Hayahsi, T.; Endo, M.; Torres, R.M.; Terrones, M. Efficient anchorage of Pt clusters on N-doped carbon nanotubes and their catalytic activity. Chem. Phys. Lett. 2008, 463, 124–129. [Google Scholar] [CrossRef]
- Kim, B.; Sigmund, W.M. Functionalized multiwall carbon nanotube/gold nanoparticle composites. Langmuir 2004, 20, 8239–8242. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, A.; Elías, A.L.; Rodríguez-Manzo, J.A.; López-Urías, F.; Rodríguez-Gattorno, G.; Lupo, F.; Rühle, M.; Smith, D.J.; Terrones, H.; Díaz, D.; et al. Efficient anchoring of silver nanoparticles on n-doped carbon nanotubes. Small 2006, 2, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.K.; Sullivan, D.I.; Turner, C.H. Characterizing the interaction of Pt and PtRu clusters with boron-doped, nitrogen-doped, and activated carbon: Density functional theory calculations and parameterization. J. Phys. Chem. C 2008, 112, 13607–13622. [Google Scholar] [CrossRef]
- Li, Y.H.; Hung, T.H.; Chen, C.W. A first-principles study of nitrogen- and boron-assisted platinum adsorption on carbon nanotubes. Carbon 2009, 47, 850–855. [Google Scholar] [CrossRef]
- An, W.; Turner, C. Chemisorption of transition-metal atoms on boron-and nitrogen-doped carbon nanotubes: Energetics and geometric and electronic structures. J. Phys. Chem. C 2009, 113, 7069–7078. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Xie, Z.; Yi, Y.; Wowsnick, G.; Sun, L.L.; Hermann, K.E.; Friedrich, M.; Kast, P.; Hävecker, M.; et al. Nature of the N-Pd interaction in nitrogen-doped carbon nanotube catalysts. ACS Catal. 2015, 5, 2740–2753. [Google Scholar] [CrossRef]
- Kabir, S.; Artyushkova, K.; Kiefer, B.; Atanassov, P. Computational and experimental evidence for a new TM–N 3/C moiety family in non-PGM electrocatalysts. Phys. Chem. Chem. Phys. 2015, 17, 17785–17789. [Google Scholar] [CrossRef] [PubMed]
- Artyushkova, K.; Kiefer, B.; Halevi, B.; Knop-Gericke, A.; Schlogl, R.; Atanassov, P. Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures. Chem. Commun. 2013, 49, 2539–2541. [Google Scholar] [CrossRef] [PubMed]
- Bulushev, D.A.; Zacharska, M.; Shlyakhova, E.V.; Chuvilin, A.L.; Guo, Y.; Beloshapkin, S.; Okotrub, A.V.; Bulusheva, L.G. Single Isolated Pd2+ Cations Supported on N-Doped Carbon as Active Sites for Hydrogen Production from Formic Acid Decomposition. ACS Catal. 2016, 6, 681–691. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2000; Volume 45, pp. 71–129. [Google Scholar]
- Feng, H.; Ma, J.; Hu, Z. Nitrogen-doped carbon nanotubes functionalized by transition metal atoms: A density functional study. J. Mater. Chem. 2010, 20, 1702–1708. [Google Scholar] [CrossRef]
- Efremenko, I.; Sheintuch, M. Carbon-supported palladium catalysts. Molecular orbital study. J. Catal. 2003, 214, 53–67. [Google Scholar] [CrossRef]
- Tang, D.; Sun, X.; Zhao, D.; Zhu, J.; Zhang, W.; Xu, X.; Zhao, Z. Nitrogen-Doped Carbon Xerogels Supporting Palladium Nanoparticles for Selective Hydrogenation Reactions: The Role of Pyridine Nitrogen Species. ChemCatChem 2018, 10, 1291–1299. [Google Scholar] [CrossRef]
- Jiang, B.; Song, S.; Wang, J.; Xie, Y.; Chu, W.; Li, H.; Xu, H.; Tian, C.; Fu, H. Nitrogen-doped graphene supported Pd@PdO core-shell clusters for C-C coupling reactions. Nano Res. 2014, 7, 1280–1290. [Google Scholar] [CrossRef]
- Rao, R.G.; Blume, R.; Hansen, T.W.; Fuentes, E.; Dreyer, K.; Moldovan, S.; Ersen, O.; Hibbitts, D.D.; Chabal, Y.J.; Schlögl, R.; et al. Interfacial charge distributions in carbon-supported palladium catalysts. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.J.; Hou, T.Z.; Zhang, Q.; Huang, J.Q.; Cheng, X.B.; Guo, M.Q.; Yuan, Z.; He, L.Y.; Wei, F. Strongly coupled interfaces between a heterogeneous carbon host and a sulfur-containing guest for highly stable lithium-sulfur batteries: Mechanistic insight into capacity degradation. Adv. Mater. Interfaces 2014, 1, 1–10. [Google Scholar] [CrossRef]
- Kim, G.; Jhi, S.H.; Park, N. Effective metal dispersion in pyridinelike nitrogen doped graphenes for hydrogen storage. Appl. Phys. Lett. 2008, 92, 2006–2009. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Zacharska, M.; Lisitsyn, A.S.; Podyacheva, O.Y.; Hage, F.S.; Ramasse, Q.M.; Bangert, U.; Bulusheva, L.G. Single Atoms of Pt-Group Metals Stabilized by N-Doped Carbon Nanofibers for Efficient Hydrogen Production from Formic Acid. ACS Catal. 2016, 6, 3442–3451. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Jhi, S.H. Carbon monoxide-tolerant platinum nanoparticle catalysts on defect-engineered graphene. ACS Nano 2011, 5, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Perazzolo, V.; Brandiele, R.; Durante, C.; Zerbetto, M.; Causin, V.; Rizzi, G.A.; Cerri, I.; Granozzi, G.; Gennaro, A. Density Functional Theory (DFT) and Experimental Evidences of Metal-Support Interaction in Platinum Nanoparticles Supported on Nitrogen- and Sulfur-Doped Mesoporous Carbons: Synthesis, Activity, and Stability. ACS Catal. 2018, 8, 1122–1137. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Dong, B.; Wang, H.; Yu, H.; Peng, F.; Yang, Y. Electron transfer dependent catalysis of Pt on N-doped carbon nanotubes: Effects of synthesis method on metal-support interaction. J. Catal. 2017, 348, 100–109. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Tsumori, N.; Liu, Z.; Himeda, Y.; Autrey, T.; Xu, Q.; Li, Z.; Yang, X.; Tsumori, N.; et al. Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid. ACS Catal. 2017, 7, 2720–2724. [Google Scholar]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Hävecker, M. Dynamics of Pd on nanocarbon in the direct synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chew, L.M.; Kostka, A.; Muhler, M.; Xia, W. The structural and electronic promoting effect of nitrogen-doped carbon nanotubes on supported Pd nanoparticles for selective olefin hydrogenation. Catal. Sci. Technol. 2013, 3, 1964–1971. [Google Scholar] [CrossRef]
- Bi, Q.Y.; Lin, J.D.; Liu, Y.M.; He, H.Y.; Huang, F.Q.; Cao, Y. Dehydrogenation of Formic Acid at Room Temperature: Boosting Palladium Nanoparticle Efficiency by Coupling with Pyridinic-Nitrogen-Doped Carbon. Angew. Chem. Int. Ed. 2016, 55, 11849–11853. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Jung, D.; Lee, K.; Hee, S.; Han, J.; Woo, S.; Chul, S.; Park, H.S. Electronically modified Pd catalysts supported on N-doped carbon for the dehydrogenation of formic acid. Int. J. Hydrogen Energy 2016, 41, 15453–15461. [Google Scholar] [CrossRef]
- Burgess, R.; Buono, C.; Davies, P.R.; Davies, R.J.; Legge, T.; Lai, A.; Lewis, R.; Morgan, D.J.; Robinson, N.; Willock, D.J. The functionalisation of graphite surfaces with nitric acid: Identification of functional groups and their effects on gold deposition. J. Catal. 2015, 323, 10–18. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, J.; Sun, Y.; Ning, W.; Hou, Z. Selective oxidation of glycerol over nitrogen-doped carbon nanotubes supported platinum catalyst in base-free solution. Catal. Commun. 2015, 70, 72–76. [Google Scholar] [CrossRef]
- Perini, L.; Durante, C.; Favaro, M.; Perazzolo, V.; Agnoli, S.; Schneider, O.; Granozzi, G.; Gennaro, A. Metal-Support Interaction in Platinum and Palladium Nanoparticles Loaded on Nitrogen-Doped Mesoporous Carbon for Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2015, 7, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Liu, H.; Banis, M.N.; Li, R.; Sun, X.; Sham, T.-K.; Ye, S.; Knights, S. Nitrogen Doping Effects on Carbon Nanotubes and the Origin of the Enhanced Electrocatalytic Activity of Supported Pt for Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. C 2011, 115, 3769–3776. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Swaidan, R.; Li, D.; Li, N. Enhanced methanol electro-oxidation activity of PtRu catalysts supported on heteroatom-doped carbon. Electrochim. Acta 2008, 53, 7622–7629. [Google Scholar] [CrossRef]
- Vinayan, B.P.; Ramaprabhu, S. Platinum-TM (TM = Fe, Co) alloy nanoparticles dispersed nitrogen doped (reduced graphene oxide-multiwalled carbon nanotube) hybrid structure cathode electrocatalysts for high performance PEMFC applications. Nanoscale 2013, 5, 5109–5118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Moyo, M.; Rayner, M.K.; Jewell, L.L.; Billing, D.G.; Coville, N.J. Autoreduction and Catalytic Performance of a Cobalt Fischer-Tropsch Synthesis Catalyst Supported on Nitrogen-Doped Carbon Spheres. ChemCatChem 2010, 2, 514–518. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Cheng, H.; Li, N.; Ma, T.Y.; Su, Y.Z. ZnCo2O4 Quantum Dots Anchored on Nitrogen-Doped Carbon Nanotubes as Reversible Oxygen Reduction/Evolution Electrocatalysts. Adv. Mater. 2016, 28, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Melke, J.; Peter, B.; Habereder, A.; Ziegler, J.; Fasel, C.; Nefedov, A.; Sezen, H.; Wöll, C.; Ehrenberg, H.; Roth, C. Metal-Support Interactions of Platinum Nanoparticles Decorated N-Doped Carbon Nanofibers for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2016, 8, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Hu, Y.; Regier, T. Chemical interaction and imaging of single Co3O4/graphene sheets studied by scanning transmission X-ray microscopy and X-ray absorption spectroscopy. Energy Environ. Sci. 2013, 6, 926–934. [Google Scholar] [CrossRef]
- Zhang, B.; Su, D.S. Probing the Metal-Support Interaction in Carbon-Supported Catalysts by using Electron Microscopy. ChemCatChem 2015, 7, 3639–3645. [Google Scholar] [CrossRef]
- Suarez-Martinez, I.; Bittencourt, C.; Ke, X.; Felten, A.; Pireaux, J.J.; Ghijsen, J.; Drube, W.; Van Tendeloo, G.; Ewels, C.P. Probing the interaction between gold nanoparticles and oxygen functionalized carbon nanotubes. Carbon 2009, 47, 1549–1554. [Google Scholar] [CrossRef]
- Yoon, H.; Ko, S.; Jang, J. Nitrogen-doped magnetic carbon nanoparticles as catalyst supports for efficient recovery and recycling. Chem. Commun. 2007, 1468–1470. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yang, F.; Rasouli, S.; Qiu, Y.; Li, Z.F.; Uzunoglu, A.; Sun, C.J.; Liu, Y.; Ferreira, P.; Li, W.; et al. Understanding Pt Nanoparticle Anchoring on Graphene Supports through Surface Functionalization. ACS Catal. 2016, 6, 2642–2653. [Google Scholar] [CrossRef]
- He, D.; Jiang, Y.; Lv, H.; Pan, M.; Mu, S. Nitrogen-doped reduced graphene oxide supports for noble metal catalysts with greatly enhanced activity and stability. Appl. Catal. B Environ. 2013, 132–133, 379–388. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Hu, H.; Wang, Y.; Chen, H.; Lou, X.W.D. Ultrathin MoS2 Nanosheets Supported on N-doped Carbon Nanoboxes with Enhanced Lithium Storage and Electrocatalytic Properties. Angew. Chem. Int. Ed. 2015, 54, 7395–7398. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Dai, C.; Wang, D.; Li, D.; Li, N. Nitrogen-doped magnetic onion-like carbon as support for Pt particles in a hybrid cathode catalyst for fuel cells. J. Mater. Chem. 2010, 20, 3059–3068. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P. Selective oxidation of alcohols and aldehydes on metal catalysts. Catal. Today 2000, 57, 127–141. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Hutchings, G.J. Selective liquid phase oxidation with supported metal nanoparticles. Chem. Sci. 2012, 3, 20–44. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Spontoni, P.; Arrigo, R.; Su, D.; Prati, L. Nitrogen functionalized carbon nanostructures supported Pd and Au–Pd NPs as catalyst for alcohols oxidation. Catal. Today 2010, 157, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Prati, L.; Villa, A.; Chan-Thaw, C.E.; Arrigo, R.; Wang, D.; Su, D.S. Gold catalyzed liquid phase oxidation of alcohol: The issue of selectivity. Faraday Discuss. 2011, 152, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shang, J.-H.; Chen, Y.; Wang, Y.; Li, Y.-X. Palladium nanoparticles supported on mesoporous carbon nitride for efficiently selective oxidation of benzyl alcohol with molecular oxygen. Appl. Catal. A Gen. 2017, 542, 380–388. [Google Scholar] [CrossRef]
- Chan-Thaw, C.E.; Villa, A.; Veith, G.M.; Prati, L. Identifying the Role of N-Heteroatom Location in the Activity of Metal Catalysts for Alcohol Oxidation. ChemCatChem 2015, 7, 1338–1346. [Google Scholar] [CrossRef]
- Ravat, V.; Nongwe, I.; Coville, N.J. Palladium-Supported Boron-Doped Hollow Carbon Spheres as Catalysts for the Solvent-free Aerobic Oxidation of Alcohols. ChemCatChem 2012, 4, 1930–1934. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, H.; Luo, Y.; Zhang, S.; Gao, J.; Xu, J. Activation of Molecular Oxygen Using Durable Cobalt Encapsulated with Nitrogen-Doped Graphitic Carbon Shells for Aerobic Oxidation of Lignin-Derived Alcohols. Chem. Eur. J. 2018, 24, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhang, K.-X.; Zhang, B.; Wang, H.-H.; Yu, Q.-Y.; Li, X.-H.; Antonietti, M.; Chen, J.-S. Activating Cobalt Nanoparticles via the Mott-Schottky Effect in Nitrogen-Rich Carbon Shells for Base-Free Aerobic Oxidation of Alcohols to Esters. J. Am. Chem. Soc. 2017, 139, 811–818. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | TOF (1/h) | Conversion (%) | Solvent | Alcohol/Metal (mol %) | T (°C) | pO2 (ATM) | Sel to Benzal-Dehyde | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pd/PR24-PS | 7260 | n.d. | solventless | 35,000 | 120 | 1.5 | 62 | [101] |
| 2 | Pd/N-PR24-PS | 65,876 | n.d. | solventless | 35,000 | 120 | 1.5 | 64 | [101] |
| 3 | AuPd/PR24-PS | 6076 | n.d. | solventless | 35,000 | 120 | 1.5 | 74 | [101] |
| 4 | AuPd/N-PR24-PS | 52,638 | n.d. | solventless | 35,000 | 120 | 1.5 | 75 | [101] |
| 5 | Pd/NCNT873K | 11,457 | 95 (after 1 h) | solventless | 3000 | 80 | 2 | 76 | [52] |
| 6 | Pd/NCNT673K | 7865 | 88 (after 1 h) | solventless | 3000 | 80 | 2 | 73 | [52] |
| 7 | Pd/NCNT473K | 2387 | 35 (after 1 h) | solventless | 3000 | 80 | 2 | 68 | [52] |
| 8 | Pd/C | n.d. | 20 (after 5 h) | solventless | n.d. | 125 | 1 | 99 | [105] |
| 9 | Pd/B-C | n.d. | >99 (after 5 h) | solventless | n.d. | 125 | 1 | 99 | [105] |
| 10 | [email protected] | n.d. | 3 (after 1 h) | CH3OH | 180 | 60 | 1 | 12 | [107] |
| 11 | Co@NC-2 | n.d. | 31 (after 1 h) | CH3OH | 180 | 60 | 1 | 52 | [107] |
| 12 | Co@NC-4 | n.d. | 58 (after 1 h) | CH3OH | 180 | 60 | 1 | 77 | [107] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campisi, S.; Chan-Thaw, C.E.; Villa, A. Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review. Appl. Sci. 2018, 8, 1159. https://doi.org/10.3390/app8071159
Campisi S, Chan-Thaw CE, Villa A. Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review. Applied Sciences. 2018; 8(7):1159. https://doi.org/10.3390/app8071159
Chicago/Turabian StyleCampisi, Sebastiano, Carine Edith Chan-Thaw, and Alberto Villa. 2018. "Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review" Applied Sciences 8, no. 7: 1159. https://doi.org/10.3390/app8071159
APA StyleCampisi, S., Chan-Thaw, C. E., & Villa, A. (2018). Understanding Heteroatom-Mediated Metal–Support Interactions in Functionalized Carbons: A Perspective Review. Applied Sciences, 8(7), 1159. https://doi.org/10.3390/app8071159