Atomic Force Spectroscopy on Ionic Liquids




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
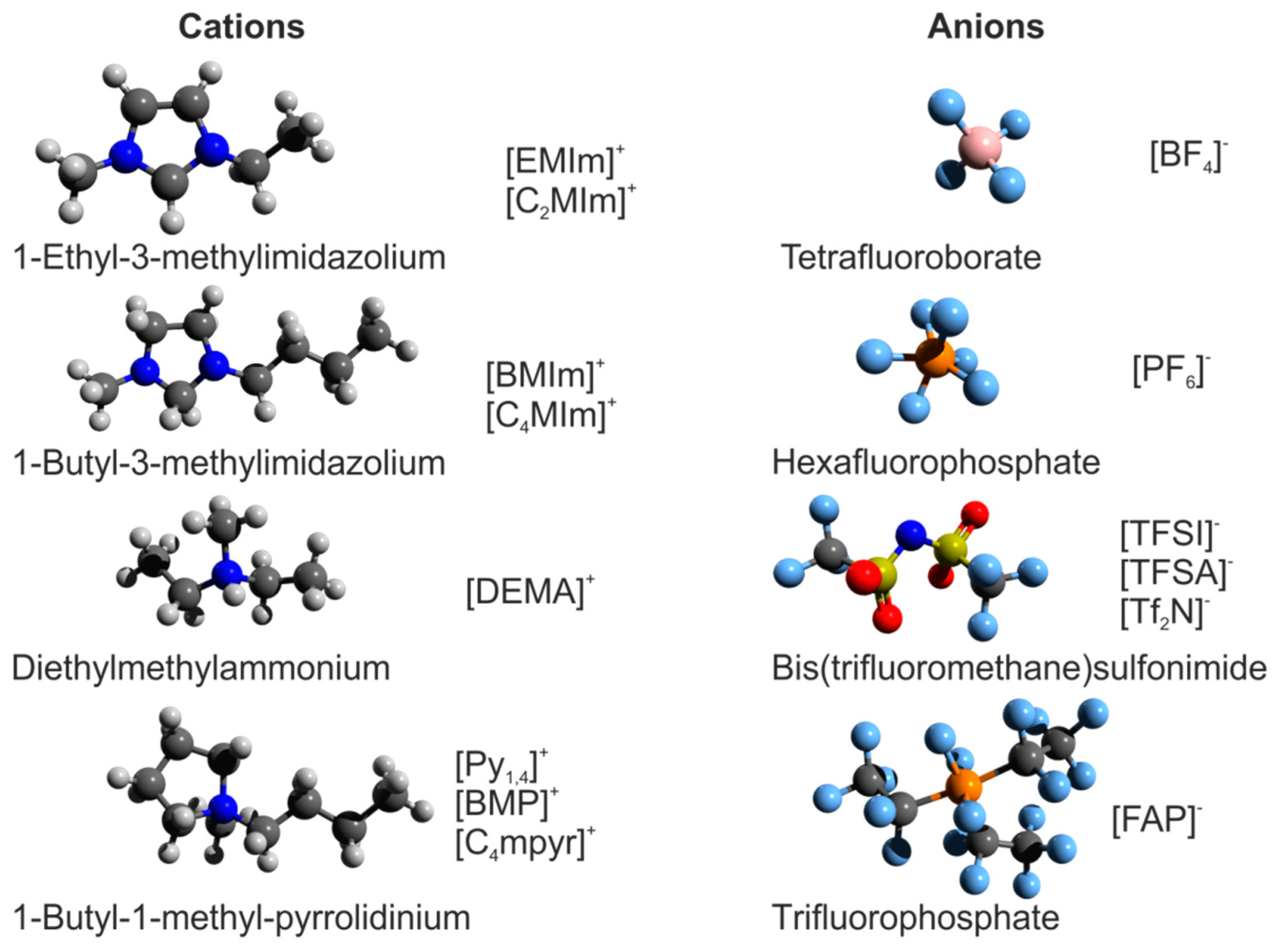
2. Bulk Structure of Ionic Liquids
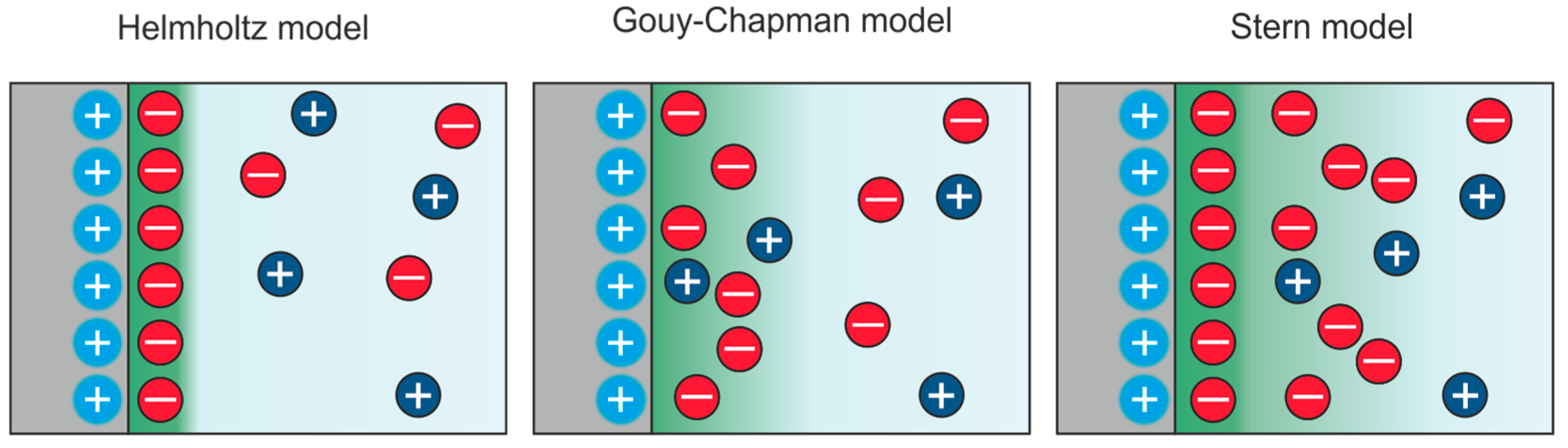
3. The Electric Double Layer at Solid-Liquid Interfaces
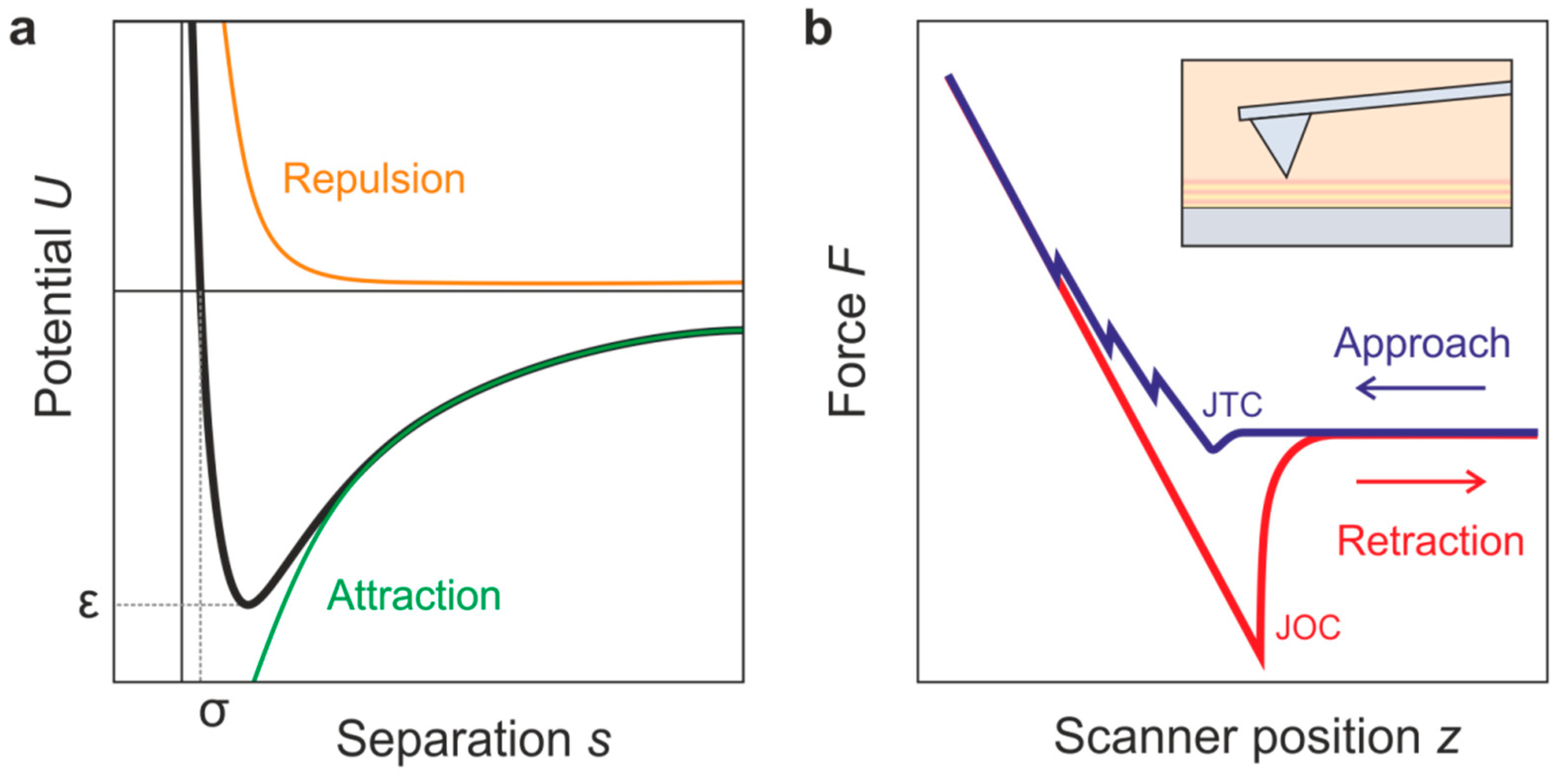
4. Principles of Atomic Force Spectroscopy
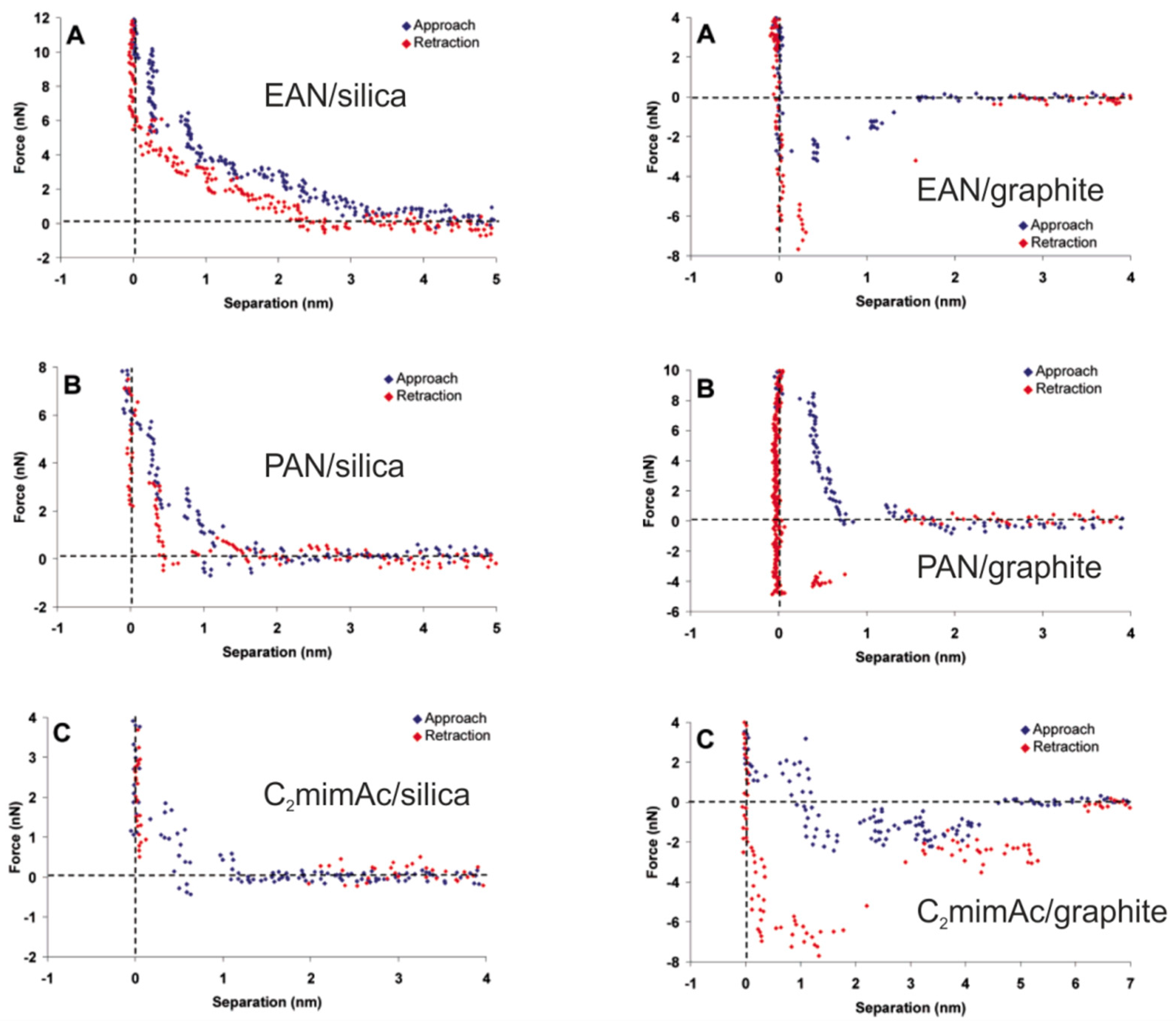
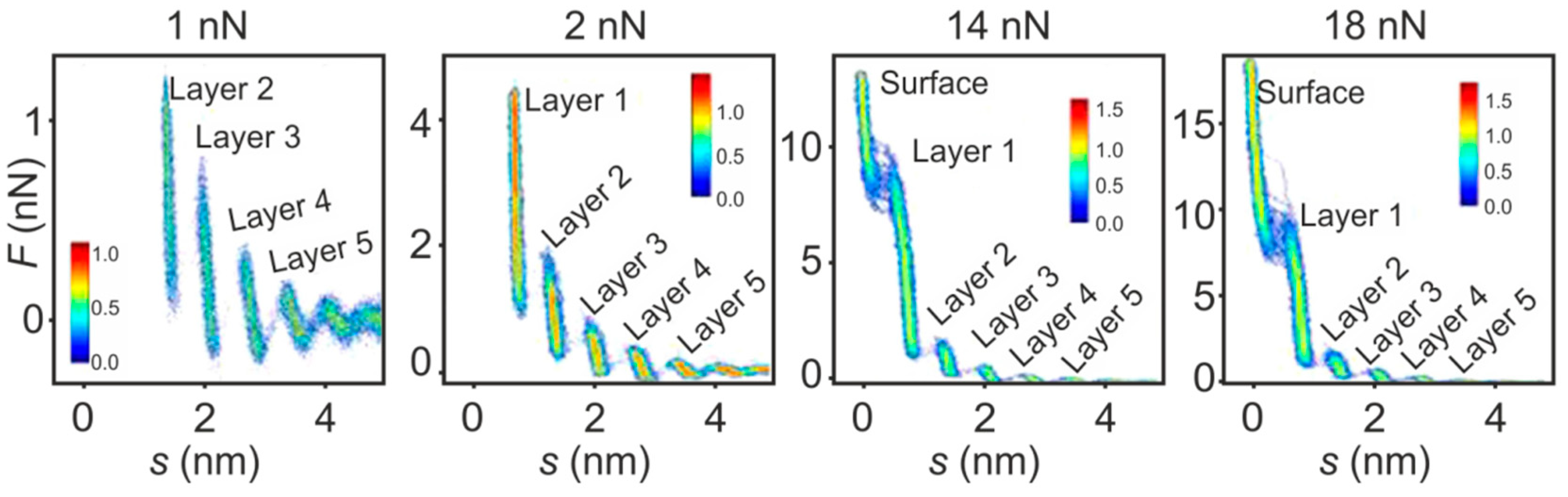
5. Structure of the Interface Layer of Ionic Liquids Revealed by AFS
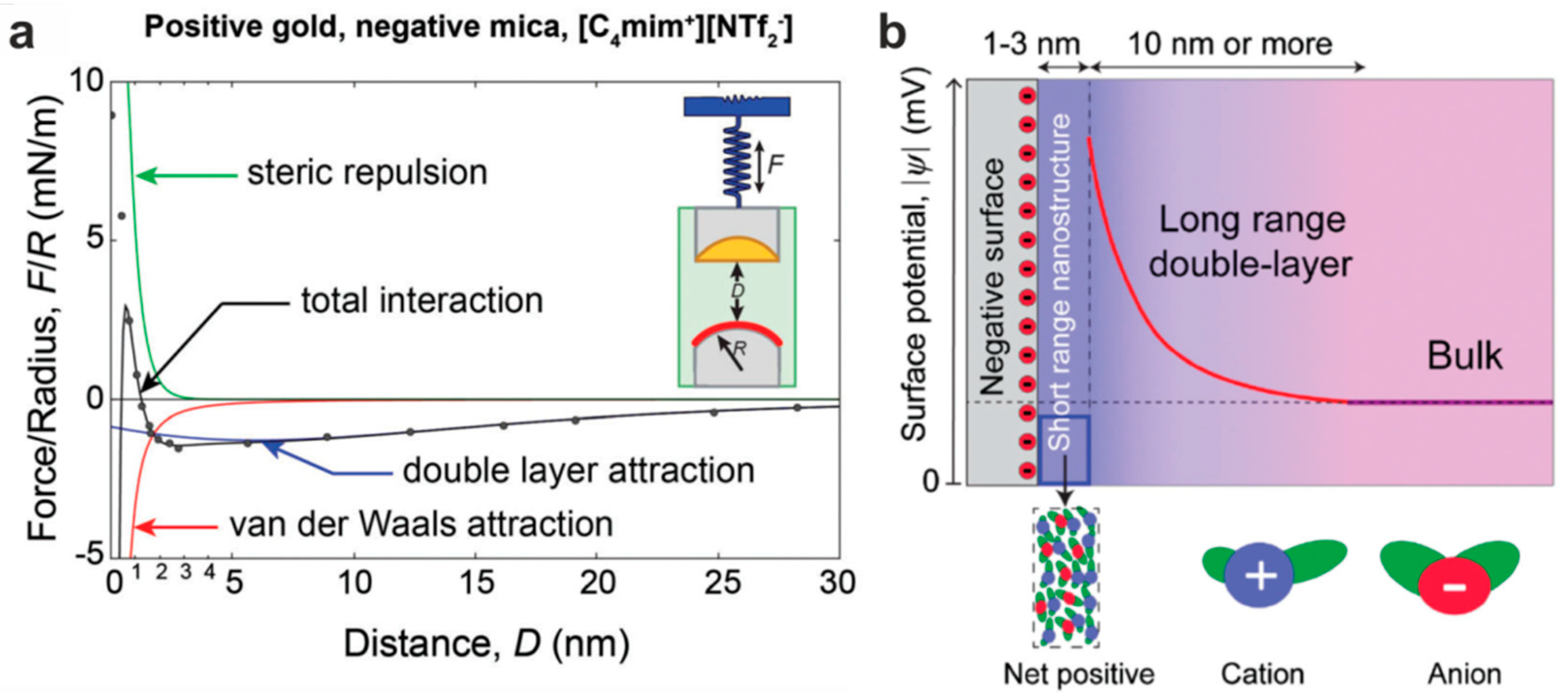
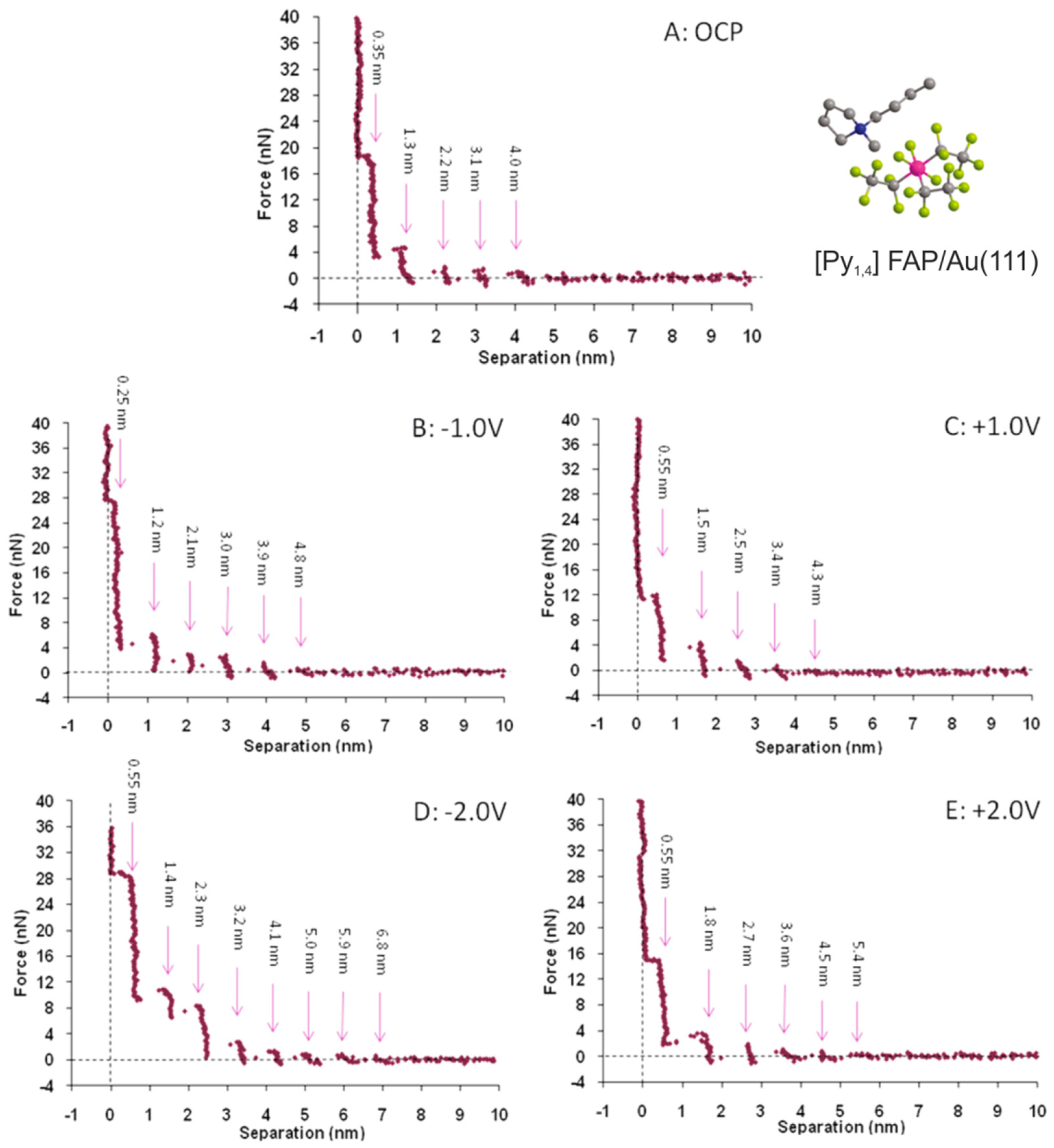
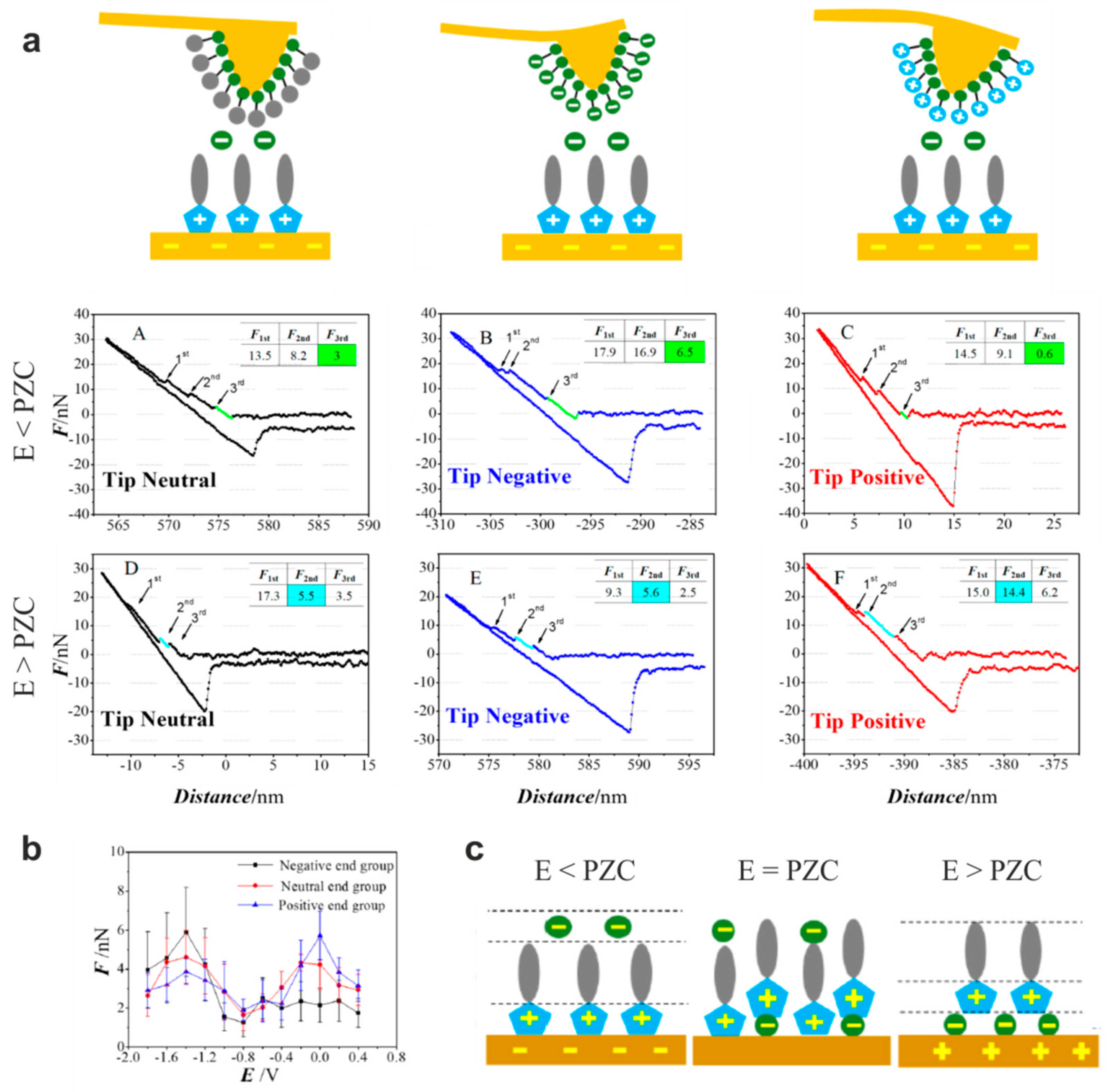
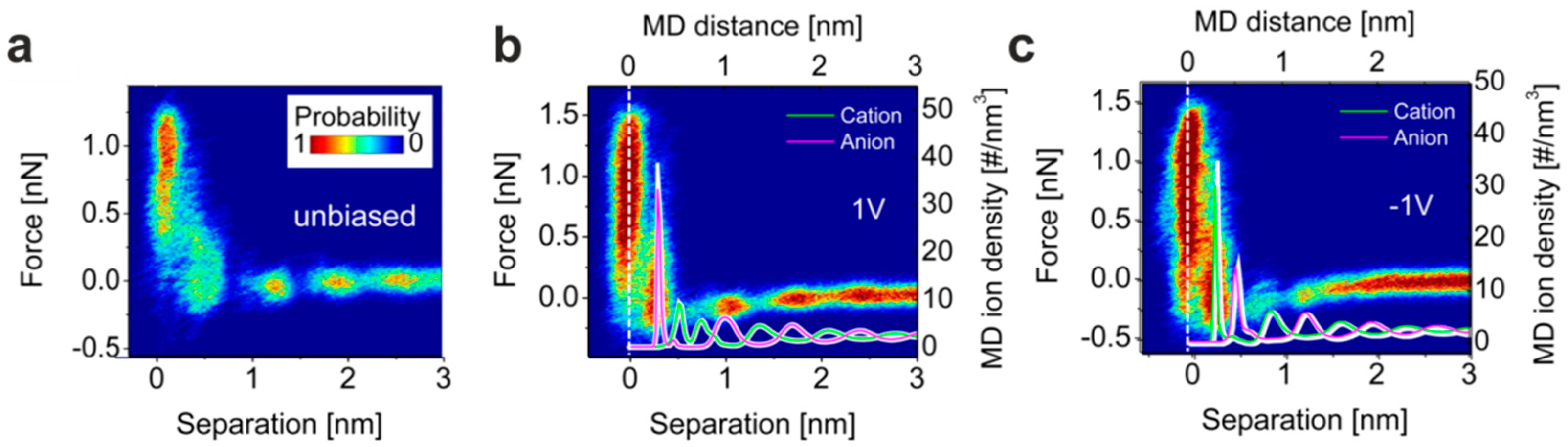
5.1. The Electrified Interface
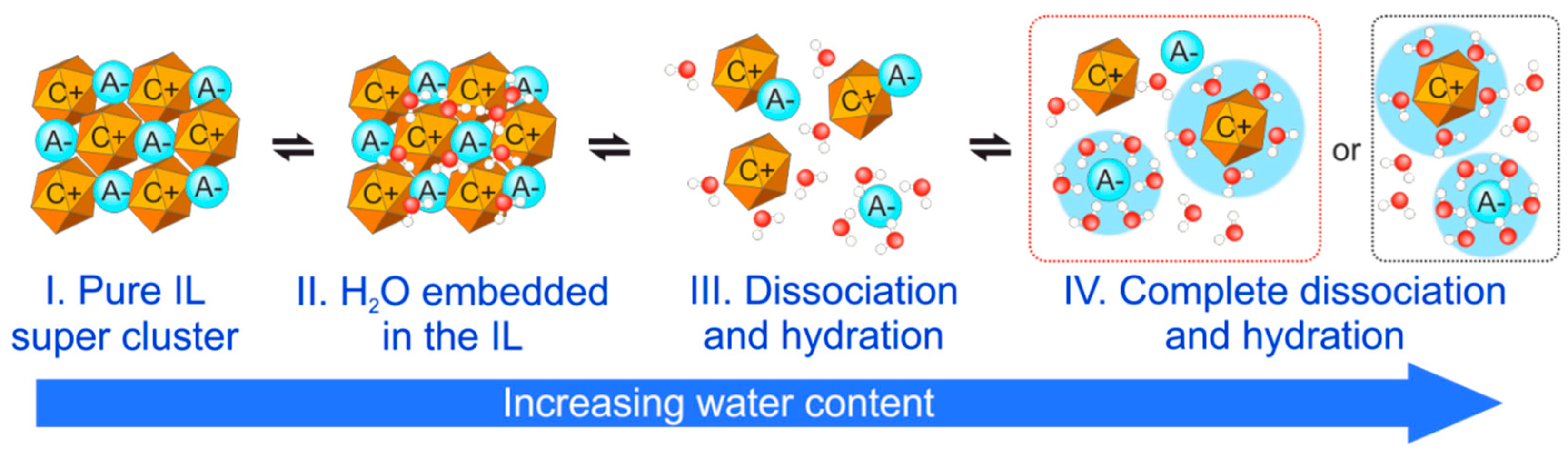
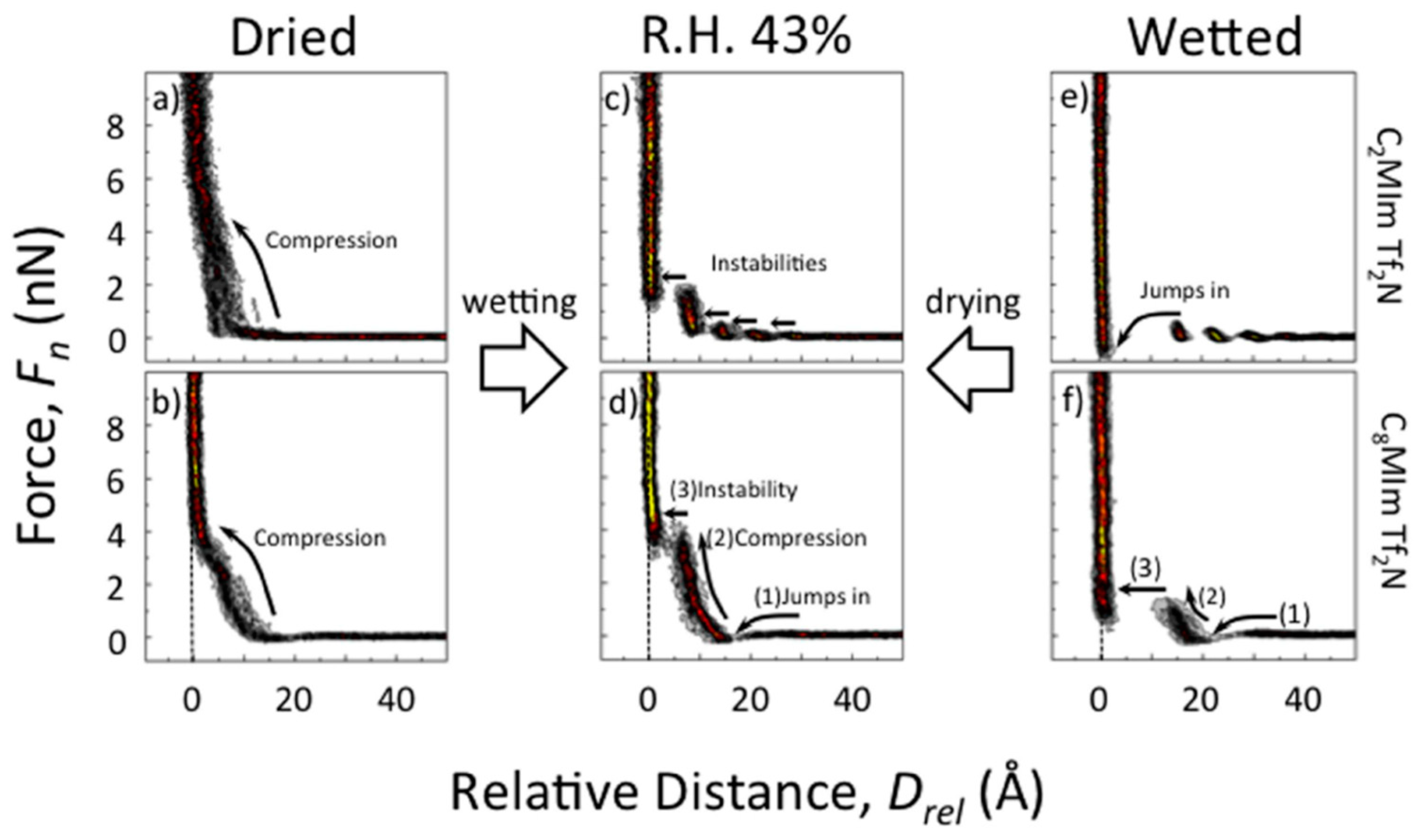
5.2. Influence of Water
5.3. Addition of Metallic Solvates
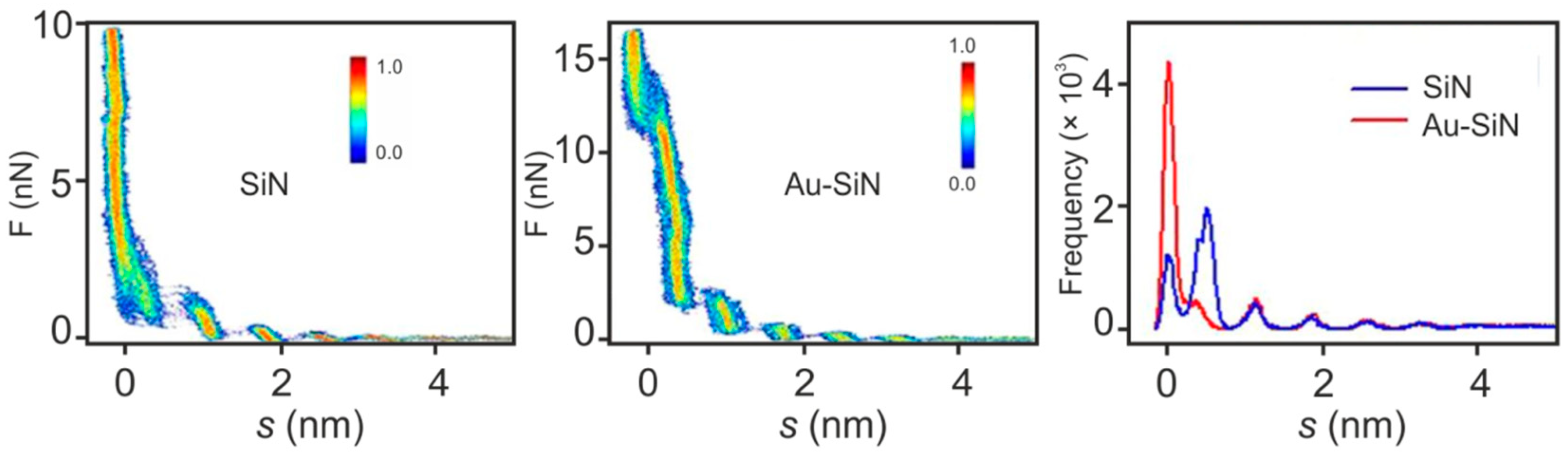
6. Towards Three-Dimensional Mapping of the Interface Layer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacFarlane, D.R.; Kar, M.; Pringle, J.M. Fundamentals of Ionic Liquids: From Chemistry to Applications; Wiley-VCH: Weinheim, Germany, 2017; ISBN 9783527339990. [Google Scholar]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef]
- Tucker, T.G.; Davidowski, S.K.; Angell, C.A. Inorganic vs Organic Cation Ionic Liquids and Their Solutions with Alkali Metal Containing Ionic Liquids. J. Electrochem. Soc. 2017, 164, H153–H158. [Google Scholar] [CrossRef] [Green Version]
- Seddon, K.R. Ionic Liquids for Clean Technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. [Google Scholar] [CrossRef]
- Freemantle, M. Designer Solvents. Chem. Eng. News 1998, 76, 32–37. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef]
- Rogers, R.D. Reflections on ionic liquids. Nature 2007, 447, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.D. The Bakerian Lecture, 1962 The structure of liquids. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1964, 280, 299–322. [Google Scholar]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [Green Version]
- Parsons, R. Models of the Electrical Double Layer. In Trends in Interfacial Electrochemistry; Springer: Dordrecht, The Netherlands, 1986; pp. 373–385. [Google Scholar]
- Kornyshev, A.A. Double-Layer in Ionic Liquids: Paradigm Change? J. Phys. Chem. B 2007, 111, 5545–5557. [Google Scholar] [CrossRef]
- Macdonald, D.D. Reflections on the history of electrochemical impedance spectroscopy. Electrochim. Acta 2006, 51, 1376–1388. [Google Scholar] [CrossRef]
- Baldelli, S. Probing Electric Fields at the Ionic Liquid–Electrode Interface Using Sum Frequency Generation Spectroscopy and Electrochemistry. J. Phys. Chem. B 2005, 109, 13049–13051. [Google Scholar] [CrossRef]
- Talaty, E.R.; Raja, S.; Storhaug, V.J.; Dölle, A.; Carper, W.R. Raman and Infrared Spectra and ab Initio Calculations of C2-4MIM Imidazolium Hexafluorophosphate Ionic Liquids. J. Phys. Chem. B 2004, 108, 13177–13184. [Google Scholar] [CrossRef]
- Mezger, M.; Schramm, S.; Schröder, H.; Reichert, H.; Deutsch, M.; De Souza, E.J.; Okasinski, J.S.; Ocko, B.M.; Honkimäki, V.; Dosch, H. Layering of [BMIM]+-based ionic liquids at a charged sapphire interface. J. Chem. Phys. 2009, 131, 094701. [Google Scholar] [CrossRef]
- Lauw, Y.; Rodopoulos, T.; Gross, M.; Nelson, A.; Gardner, R.; Horne, M.D. Electrochemical cell for neutron reflectometry studies of the structure of ionic liquids at electrified interface. Rev. Sci. Instrum. 2010, 81, 074101. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, P.; Wang, Y.; Kan, Y.; Chen, Y. Surface force apparatus studies on the surface interaction of [Cnmim+][BF4−] and [Cnmim+][PF6−] ionic liquids. In Proceedings of the 2017 IEEE International Conference on Manipulation, Manufacturing and Measurement on the Nanoscale (3M-NANO), Shanghai, China, 7–11 August 2017; pp. 300–304. [Google Scholar]
- Black, J.M.; Zhu, M.; Zhang, P.; Unocic, R.R.; Guo, D.; Okatan, M.B.; Dai, S.; Cummings, P.T.; Kalinin, S.V.; Feng, G.; et al. Fundamental aspects of electric double layer force-distance measurements at liquid-solid interfaces using atomic force microscopy. Sci. Rep. 2016, 6, 32389. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.A.; Pádua, A.A.H. Nanostructural Organization in Ionic Liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [Google Scholar] [CrossRef]
- Araque, J.C.; Hettige, J.J.; Margulis, C.J. Modern Room Temperature Ionic Liquids, a Simple Guide to Understanding Their Structure and How It May Relate to Dynamics. J. Phys. Chem. B 2015, 119, 12727–12740. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J. On the solid, liquid and solution structural organization of imidazolium ionic liquids. J. Braz. Chem. Soc. 2004, 15, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Triolo, A.; Russina, O.; Bleif, H.-J.; Di Cola, E. Nanoscale Segregation in Room Temperature Ionic Liquids. J. Phys. Chem. B 2007, 111, 4641–4644. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J. From Molten Salts to Ionic Liquids: A “Nano” Journey. Acc. Chem. Res. 2011, 44, 1223–1231. [Google Scholar] [CrossRef]
- Shimizu, K.; Tariq, M.; Freitas, A.A.; Pádua, A.A.H.; Lopes, J.N.C.; Shimizu, K.; Tariq, M.; Freitas, A.A.; Pádua, A.A.H.; Lopes, J.N.C. Self-Organization in Ionic Liquids: From Bulk to Interfaces and Films. J. Braz. Chem. Soc. 2015, 27, 349–362. [Google Scholar] [CrossRef]
- Hardacre, C.; Holbrey, J.D.; Nieuwenhuyzen, M.; Youngs, T.G.A. Structure and Solvation in Ionic Liquids. Acc. Chem. Res. 2007, 40, 1146–1155. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, W.; Yan, T.; Voth, G.A. Understanding Ionic Liquids through Atomistic and Coarse-Grained Molecular Dynamics Simulations. Acc. Chem. Res. 2007, 40, 1193–1199. [Google Scholar] [CrossRef]
- Greaves, T.L.; Ha, K.; Muir, B.W.; Howard, S.C.; Weerawardena, A.; Kirby, N.; Drummond, C.J. Protic ionic liquids (PILs) nanostructure and physicochemical properties: Development of high-throughput methodology for PIL creation and property screens. Phys. Chem. Chem. Phys. 2015, 17, 2357–2365. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Evolving Structure–Property Relationships and Expanding Applications. Chem. Rev. 2015, 115, 11379–11448. [Google Scholar] [CrossRef]
- Henderson, W.A.; Fylstra, P.; De Long, H.C.; Trulove, P.C.; Parsons, S. Crystal structure of the ionic liquid EtNH3NO3—Insights into the thermal phase behavior of protic ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 16041. [Google Scholar] [CrossRef]
- Lo Celso, F.; Appetecchi, G.B.; Jafta, C.J.; Gontrani, L.; Canongia Lopes, J.N.; Triolo, A.; Russina, O. Nanoscale organization in the fluorinated room temperature ionic liquid: Tetraethyl ammonium (trifluoromethanesulfonyl)(nonafluorobutylsulfonyl)imide. J. Chem. Phys. 2018, 148, 193816. [Google Scholar] [CrossRef] [PubMed]
- Russina, O.; Lo Celso, F.; Di Michiel, M.; Passerini, S.; Appetecchi, G.B.; Castiglione, F.; Mele, A.; Caminiti, R.; Triolo, A. Mesoscopic structural organization in triphilic room temperature ionic liquids. Faraday Discuss. 2013, 167, 499. [Google Scholar] [CrossRef] [PubMed]
- Hettige, J.J.; Araque, J.C.; Margulis, C.J. Bicontinuity and Multiple Length Scale Ordering in Triphilic Hydrogen-Bonding Ionic Liquids. J. Phys. Chem. B 2014, 118, 12706–12716. [Google Scholar] [CrossRef]
- Seddon, K.R.; Stark, A.; Torres, M.-J. Influence of chloride, water, and organic solvents on the physical properties of ionic liquids. Pure Appl. Chem. 2000, 72, 2275–2287. [Google Scholar] [CrossRef]
- Chatel, G.; Pereira, J.F.B.; Debbeti, V.; Wang, H.; Rogers, R.D. Mixing ionic liquids—“simple mixtures” or “double salts”? Green Chem. 2014, 16, 2051. [Google Scholar] [CrossRef]
- Yaghini, N.; Pitawala, J.; Matic, A.; Martinelli, A. Effect of Water on the Local Structure and Phase Behavior of Imidazolium-Based Protic Ionic Liquids. J. Phys. Chem. B 2015, 119, 1611–1622. [Google Scholar] [CrossRef]
- Niazi, A.A.; Rabideau, B.D.; Ismail, A.E. Effects of Water Concentration on the Structural and Diffusion Properties of Imidazolium-Based Ionic Liquid–Water Mixtures. J. Phys. Chem. B 2013, 117, 1378–1388. [Google Scholar] [CrossRef]
- Martins, V.L.; Nicolau, B.G.; Urahata, S.M.; Ribeiro, M.C.C.; Torresi, R.M. Influence of the Water Content on the Structure and Physicochemical Properties of an Ionic Liquid and Its Li + Mixture. J. Phys. Chem. B 2013, 117, 8782–8792. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sun, X.; Chen, Y.; Mu, T. Water Sorption in Amino Acid Ionic Liquids: Kinetic, Mechanism, and Correlations between Hygroscopicity and Solvatochromic Parameters. ACS Sustain. Chem. Eng. 2014, 2, 138–148. [Google Scholar] [CrossRef]
- O’Mahony, A.M.; Silvester, D.S.; Aldous, L.; Hardacre, C.; Compton, R.G. Effect of Water on the Electrochemical Window and Potential Limits of Room-Temperature Ionic Liquids. J. Chem. Eng. Data 2008, 53, 2884–2891. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, Y.; Sun, X.; Zhang, Z.; Mu, T. Water sorption in ionic liquids: kinetics, mechanisms and hydrophilicity. Phys. Chem. Chem. Phys. 2012, 14, 12252. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, Y.; Lu, X.; Zhao, C.; Yan, C.; Mu, T. Water sorption in protic ionic liquids: Correlation between hygroscopicity and polarity. New J. Chem. 2013, 37, 1959. [Google Scholar] [CrossRef]
- Ma, C.; Laaksonen, A.; Liu, C.; Lu, X.; Ji, X. The peculiar effect of water on ionic liquids and deep eutectic solvents. Chem. Soc. Rev. 2018, 47, 8685–8720. [Google Scholar] [CrossRef] [Green Version]
- Blesic, M.; Marques, M.H.; Plechkova, N.V.; Seddon, K.R.; Rebelo, L.P.N.; Lopes, A. Self-aggregation of ionic liquids: Micelle formation in aqueous solution. Green Chem. 2007, 9, 481. [Google Scholar] [CrossRef]
- Bica, K.; Gärtner, P.; Gritsch, P.J.; Ressmann, A.K.; Schröder, C.; Zirbs, R. Micellar catalysis in aqueous–ionic liquid systems. Chem. Commun. 2012, 48, 5013. [Google Scholar] [CrossRef]
- Elbourne, A.; Voïtchovsky, K.; Warr, G.G.; Atkin, R. Ion structure controls ionic liquid near-surface and interfacial nanostructure. Chem. Sci. 2015, 6, 527–536. [Google Scholar] [CrossRef]
- Wippermann, K.; Wackerl, J.; Lehnert, W.; Huber, B.; Korte, C. 2-Sulfoethylammonium Trifluoromethanesulfonate as an Ionic Liquid for High Temperature PEM Fuel Cells. J. Electrochem. Soc. 2016, 163, F25–F37. [Google Scholar] [CrossRef]
- Helmholtz, H. Ueber einige Gesetze der Vertheilung elektrischer Ströme in körperlichen Leitern mit Anwendung auf die thierisch-elektrischen Versuche. Ann. der Phys. und Chemie 1853, 165, 211–233. [Google Scholar] [CrossRef]
- Gouy, M. Sur la constitution de la charge électrique à la surface d’un électrolyte. J. Phys. Théorique Appliquée 1910, 9, 457–468. [Google Scholar] [CrossRef]
- Chapman, D.L. LI. A contribution to the theory of electrocapillarity. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1913, 25, 475–481. [Google Scholar] [CrossRef]
- Stern, O. Zur Theorie der elektrolytischen Doppelschicht. Zeitschrift für Elektrochemie und Angew. Phys. Chemie 1924, 30, 508–516. [Google Scholar]
- Schmickler, W.; Henderson, D. New models for the structure of the electrochemical interface. Prog. Surf. Sci. 1986, 22, 323–419. [Google Scholar] [CrossRef]
- Schmickler, W.; Henderson, D. The interphase between jellium and a hard sphere electrolyte. A model for the electric double layer. J. Chem. Phys. 1984, 80, 3381–3386. [Google Scholar] [CrossRef]
- Badiali, J.P. Contribution of the metal to the differential capacitance of the ideally polarizable electrode. Electrochim. Acta 1986, 31, 149–154. [Google Scholar] [CrossRef]
- Fedorov, M.V.; Kornyshev, A.A. Ionic Liquids at Electrified Interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [Green Version]
- Bazant, M.Z.; Storey, B.D.; Kornyshev, A.A. Double Layer in Ionic Liquids: Overscreening versus Crowding. Phys. Rev. Lett. 2011, 106, 046102. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale Studies on Ionic Liquids. Chem. Rev. 2017, 117, 6636–6695. [Google Scholar] [CrossRef]
- Merlet, C.; Limmer, D.T.; Salanne, M.; van Roij, R.; Madden, P.A.; Chandler, D.; Rotenberg, B. The Electric Double Layer Has a Life of its Own. J. Phys. Chem. C 2014, 118, 18291–18298. [Google Scholar] [CrossRef]
- Fedorov, M.V.; Georgi, N.; Kornyshev, A.A. Double layer in ionic liquids: The nature of the camel shape of capacitance. Electrochem. Commun. 2010, 12, 296–299. [Google Scholar] [CrossRef]
- Gebbie, M.A.; Smith, A.M.; Dobbs, H.A.; Lee, A.A.; Warr, G.G.; Banquy, X.; Valtiner, M.; Rutland, M.W.; Israelachvili, J.N.; Perkin, S.; et al. Long range electrostatic forces in ionic liquids. Chem. Commun. 2017, 53, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic Force Microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [Green Version]
- Cappella, B.; Dietler, G. Force-distance curves by atomic force microscopy. Surf. Sci. Rep. 1999, 34, 1–104. [Google Scholar] [CrossRef] [Green Version]
- Schimmel, T.; Koch, T.; Küppers, J.; Lux-Steiner, M. True atomic resolution under ambient conditions obtained by atomic force microscopy in the contact mode. Appl. Phys. A Mater. Sci. Process. 1999, 68, 399–402. [Google Scholar] [CrossRef]
- Morita, S.; Giessibl, F.J.; Meyer, E.; Wiesendanger, R. (Eds.) Noncontact Atomic Force Microscopy; NanoScience and Technology; Springer International Publishing: Cham, The Netherlands, 2015; ISBN 978-3-319-15587-6. [Google Scholar]
- Giessibl, F.J. The qPlus sensor, a powerful core for the atomic force microscope. Rev. Sci. Instrum. 2019, 90, 011101. [Google Scholar] [CrossRef]
- Mungse, H.P.; Ichii, T.; Utsunomiya, T.; Sugimura, H. Investigation of BMI-PF6 Ionic Liquid/Graphite Interface Using Frequency Modulation Atomic Force Microscopy. MRS Adv. 2018, 3, 2725–2733. [Google Scholar] [CrossRef]
- Dufrêne, Y.F.; Ando, T.; Garcia, R.; Alsteens, D.; Martinez-Martin, D.; Engel, A.; Gerber, C.; Müller, D.J. Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat. Nanotechnol. 2017, 12, 295–307. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Wu, G.; Hu, J. Coexistence of Liquid and Solid Phases of Bmim-PF6 Ionic Liquid on Mica Surfaces at Room Temperature. J. Am. Chem. Soc. 2006, 128, 7456–7457. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Liu, H.; Li, Z.; Li, J. Electrodeposition of Platinum in Room-Temperature Ionic Liquids and Electrocatalytic Effect on Electro-oxidation of Methanol. J. Electrochem. Soc. 2005, 152, E146. [Google Scholar] [CrossRef]
- Atkin, R.; Warr, G.G. Self-Assembly of a Nonionic Surfactant at the Graphite/Ionic Liquid Interface. J. Am. Chem. Soc. 2005, 127, 11940–11941. [Google Scholar] [CrossRef]
- Kubo, K.; Hirai, N.; Tanaka, T.; Hara, S. Decay of nano-islands located on Au(1 0 0) electrode in room temperature molten salt (EMImBF4). Surf. Sci. 2004, 565, L271–L276. [Google Scholar] [CrossRef]
- Kubo, K.; Hirai, N.; Tanaka, T.; Hara, S. In situ observation on Au(1 0 0) surface in molten EMImBF4 by electrochemical atomic force microscopy (EC-AFM). Surf. Sci. 2003, 546, L785–L788. [Google Scholar] [CrossRef]
- Seo, Y.; Jhe, W. Atomic force microscopy and spectroscopy. Rep. Prog. Phys. 2008, 71, 016101. [Google Scholar] [CrossRef]
- Takeyasu, K. (Ed.) Atomic Force Microscopy in Nanobiology; Jenny Stanford Publishing: Singapore, 2016; ISBN 9780429074219. [Google Scholar]
- Trewby, W.; Livesey, D.; Voïtchovsky, K. Buffering agents modify the hydration landscape at charged interfaces. Soft Matter 2016, 12, 2642–2651. [Google Scholar] [CrossRef] [Green Version]
- Senden, T.J. Force microscopy and surface interactions. Curr. Opin. Colloid Interface Sci. 2001, 6, 95–101. [Google Scholar] [CrossRef]
- Burnham, N.A.; Colton, R.J.; Pollock, H.M. Interpretation of force curves in force microscopy. Nanotechnology 1993, 4, 64–80. [Google Scholar] [CrossRef]
- Leite, F.L.; Bueno, C.C.; Da Róz, A.L.; Ziemath, E.C.; Oliveira, O.N.; Leite, F.L.; Bueno, C.C.; Da Róz, A.L.; Ziemath, E.C.; Oliveira, O.N. Theoretical Models for Surface Forces and Adhesion and Their Measurement Using Atomic Force Microscopy. Int. J. Mol. Sci. 2012, 13, 12773–12856. [Google Scholar] [CrossRef] [PubMed]
- Perkin, S. Ionic liquids in confined geometries. Phys. Chem. Chem. Phys. 2012, 14, 5052. [Google Scholar] [CrossRef]
- Amano, K.; Yokota, Y.; Ichii, T.; Yoshida, N.; Nishi, N.; Katakura, S.; Imanishi, A.; Fukui, K.; Sakka, T. A relationship between the force curve measured by atomic force microscopy in an ionic liquid and its density distribution on a substrate. Phys. Chem. Chem. Phys. 2017, 19, 30504–30512. [Google Scholar] [CrossRef] [Green Version]
- Akrami, S.M.R.; Nakayachi, H.; Watanabe-Nakayama, T.; Asakawa, H.; Fukuma, T. Significant improvements in stability and reproducibility of atomic-scale atomic force microscopy in liquid. Nanotechnology 2014, 25, 455701. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-S.; Huefner, N.D.; Chan, W.S.; Dryden, P.; Hagenhoff, B.; Beebe, T.P. Organic and Inorganic Contamination on Commercial AFM Cantilevers. Langmuir 1999, 15, 6522–6526. [Google Scholar] [CrossRef]
- Gołek, F.; Mazur, P.; Ryszka, Z.; Zuber, S. AFM image artifacts. Appl. Surf. Sci. 2014, 304, 11–19. [Google Scholar] [CrossRef]
- Zhong, Y.-X.; Yan, J.-W.; Li, M.-G.; Zhang, X.; He, D.-W.; Mao, B.-W. Resolving Fine Structures of the Electric Double Layer of Electrochemical Interfaces in Ionic Liquids with an AFM Tip Modification Strategy. J. Am. Chem. Soc. 2014, 136, 14682–14685. [Google Scholar] [CrossRef]
- Best, R.B.; Brockwell, D.J.; Toca-Herrera, J.L.; Blake, A.W.; Smith, D.A.; Radford, S.E.; Clarke, J. Force mode atomic force microscopy as a tool for protein folding studies. Anal. Chim. Acta 2003, 479, 87–105. [Google Scholar] [CrossRef]
- Brockwell, D.J.; Paci, E.; Zinober, R.C.; Beddard, G.S.; Olmsted, P.D.; Smith, D.A.; Perham, R.N.; Radford, S.E. Pulling geometry defines the mechanical resistance of a β-sheet protein. Nat. Struct. Mol. Biol. 2003, 10, 731–737. [Google Scholar] [CrossRef]
- Atkin, R.; Warr, G.G. Structure in Confined Room-Temperature Ionic Liquids. J. Phys. Chem. C 2007, 111, 5162–5168. [Google Scholar] [CrossRef]
- Hayes, R.; Borisenko, N.; Tam, M.K.; Howlett, P.C.; Endres, F.; Atkin, R. Double Layer Structure of Ionic Liquids at the Au(111) Electrode Interface: An Atomic Force Microscopy Investigation. J. Phys. Chem. C 2011, 115, 6855–6863. [Google Scholar] [CrossRef]
- Hoth, J.; Hausen, F.; Müser, M.H.; Bennewitz, R. Force microscopy of layering and friction in an ionic liquid. J. Phys. Condens. Matter 2014, 26, 284110. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. At the interface: Solvation and designing ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 1709. [Google Scholar] [CrossRef]
- Yan, J.-W.; Tian, Z.-Q.; Mao, B.-W. Molecular-level understanding of electric double layer in ionic liquids. Curr. Opin. Electrochem. 2017, 4, 105–111. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, Y.-X.; Yan, J.-W.; Su, Y.-Z.; Zhang, M.; Mao, B.-W. Probing double layer structures of Au (111)–BMIPF6 ionic liquid interfaces from potential-dependent AFM force curves. Chem. Commun. 2012, 48, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Atkin, R.; Borisenko, N.; Drüschler, M.; Endres, F.; Hayes, R.; Huber, B.; Roling, B. Structure and dynamics of the interfacial layer between ionic liquids and electrode materials. J. Mol. Liq. 2014, 192, 44–54. [Google Scholar] [CrossRef]
- Atkin, R.; Borisenko, N.; Drüschler, M.; El Abedin, S.Z.; Endres, F.; Hayes, R.; Huber, B.; Roling, B. An in situ STM/AFM and impedance spectroscopy study of the extremely pure 1-butyl-1-methylpyrrolidinium tris(pentafluoroethyl)trifluorophosphate/Au(111) interface: Potential dependent solvation layers and the herringbone reconstruction. Phys. Chem. Chem. Phys. 2011, 13, 6849. [Google Scholar] [CrossRef]
- Atkin, R.; El Abedin, S.Z.; Hayes, R.; Gasparotto, L.H.S.; Borisenko, N.; Endres, F. AFM and STM Studies on the Surface Interaction of [BMP]TFSA and [EMIm]TFSA Ionic Liquids with Au(111). J. Phys. Chem. C 2009, 113, 13266–13272. [Google Scholar] [CrossRef]
- Liu, Z.; Cui, T.; Lu, T.; Shapouri Ghazvini, M.; Endres, F. Anion Effects on the Solid/Ionic Liquid Interface and the Electrodeposition of Zinc. J. Phys. Chem. C 2016, 120, 20224–20231. [Google Scholar] [CrossRef]
- Werzer, O.; Warr, G.G.; Atkin, R. Compact Poly(ethylene oxide) Structures Adsorbed at the Ethylammonium Nitrate–Silica Interface. Langmuir 2011, 27, 3541–3549. [Google Scholar] [CrossRef]
- Li, H.; Endres, F.; Atkin, R. Effect of alkyl chain length and anion species on the interfacial nanostructure of ionic liquids at the Au(111)–ionic liquid interface as a function of potential. Phys. Chem. Chem. Phys. 2013, 15, 14624. [Google Scholar] [CrossRef]
- Lewandowski, A.; Olejniczak, A.; Galinski, M.; Stepniak, I. Performance of carbon–carbon supercapacitors based on organic, aqueous and ionic liquid electrolytes. J. Power Sources 2010, 195, 5814–5819. [Google Scholar] [CrossRef]
- Black, J.M.; Walters, D.; Labuda, A.; Feng, G.; Hillesheim, P.C.; Dai, S.; Cummings, P.T.; Kalinin, S.V.; Proksch, R.; Balke, N. Bias-Dependent Molecular-Level Structure of Electrical Double Layer in Ionic Liquid on Graphite. Nano Lett. 2013, 13, 5954–5960. [Google Scholar] [CrossRef]
- Jurado, L.A.; Espinosa-Marzal, R.M. Insight into the Electrical Double Layer of an Ionic Liquid on Graphene. Sci. Rep. 2017, 7, 4225. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, A.; Jurado, L.A.; Ramakrishna, S.N.; Arcifa, A.; Rossi, A.; Spencer, N.D.; Espinosa-Marzal, R.M. Layering of ionic liquids on rough surfaces. Nanoscale 2016, 8, 4094–4106. [Google Scholar] [CrossRef]
- Radiom, M.; Pedraz, P.; Pilkington, G.; Rohlmann, P.; Glavatskih, S.; Rutland, M.; Radiom, M.; Pedraz, P.; Pilkington, G.; Rohlmann, P.; et al. Anomalous Interfacial Structuring of a Non-Halogenated Ionic Liquid: Effect of Substrate and Temperature. Colloids Interfaces 2018, 2, 60. [Google Scholar] [CrossRef]
- Cheng, H.-W.; Dienemann, J.-N.; Stock, P.; Merola, C.; Chen, Y.-J.; Valtiner, M. The Effect of Water and Confinement on Self-Assembly of Imidazolium Based Ionic Liquids at Mica Interfaces. Sci. Rep. 2016, 6, 30058. [Google Scholar] [CrossRef] [PubMed]
- Jurado, L.A.; Kim, H.; Rossi, A.; Arcifa, A.; Schuh, J.K.; Spencer, N.D.; Leal, C.; Ewoldt, R.H.; Espinosa-Marzal, R.M. Effect of the environmental humidity on the bulk, interfacial and nanoconfined properties of an ionic liquid. Phys. Chem. Chem. Phys. 2016, 18, 22719–22730. [Google Scholar] [CrossRef]
- Broderick, A.; Newberg, J.T. Water at Ionic Liquid Interfaces. In Ionic Liquids: Current State and Future Directions; Shiflett, M.B., Scurto, A.M., Eds.; ACS Publications: Washington, DC, USA, 2017; pp. 227–249. [Google Scholar]
- Bi, S.; Wang, R.; Liu, S.; Yan, J.; Mao, B.; Kornyshev, A.A.; Feng, G. Minimizing the electrosorption of water from humid ionic liquids on electrodes. Nat. Commun. 2018, 9, 5222. [Google Scholar] [CrossRef]
- Zhong, Y.; Yan, J.; Li, M.; Chen, L.; Mao, B. The Electric Double Layer in an Ionic Liquid Incorporated with Water Molecules: Atomic Force Microscopy Force Curve Study. ChemElectroChem 2016, 3, 2221–2226. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Atkin, R.; Priest, C. Influence of Water on the Interfacial Nanostructure and Wetting of [Rmim][NTf2] Ionic Liquids at Mica Surfaces. Langmuir 2016, 32, 8818–8825. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, F.; Zhao, C. Humidity-accelerated spreading of ionic liquids on a mica surface. RSC Adv. 2017, 7, 42718–42724. [Google Scholar] [CrossRef] [Green Version]
- Kawada, S.; Kodama, E.; Sato, K.; Ogawa, S.; Watanabe, M.; Okubo, H.; Sasaki, S. Effect of water on the interfacial structures of room-temperature ionic liquids. Surf. Interface Anal. 2019, 51, 17–20. [Google Scholar] [CrossRef]
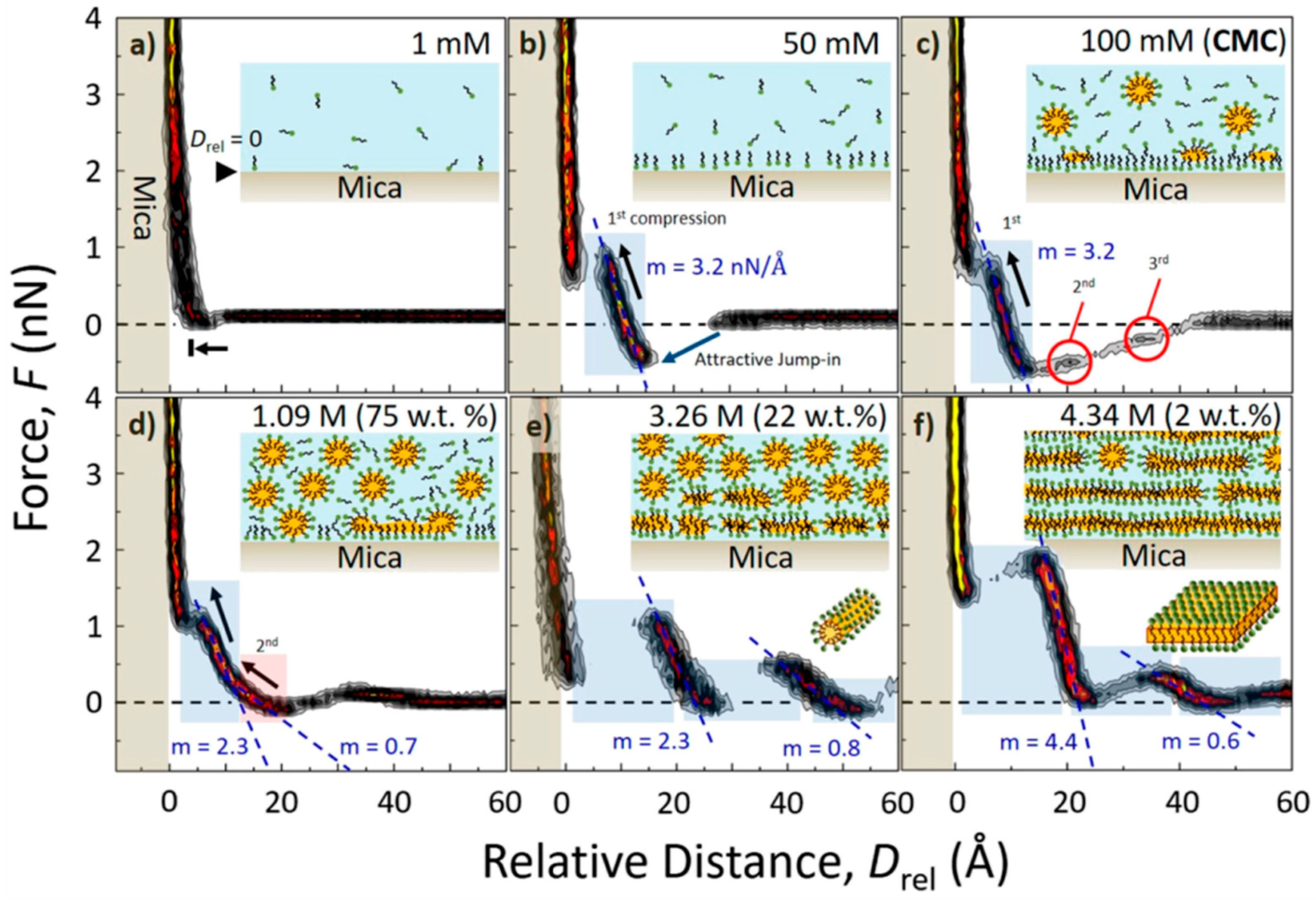
- Cheng, H.-W.; Weiss, H.; Stock, P.; Chen, Y.-J.; Reinecke, C.R.; Dienemann, J.-N.; Mezger, M.; Valtiner, M. Effect of Concentration on the Interfacial and Bulk Structure of Ionic Liquids in Aqueous Solution. Langmuir 2018, 34, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Lahiri, A.; Carstens, T.; Borisenko, N.; Pulletikurthi, G.; Kuhl, C.; Endres, F. Influence of Water on the Electrified Ionic Liquid/Solid Interface: A Direct Observation of the Transition from a Multilayered Structure to a Double-Layer Structure. J. Phys. Chem. C 2016, 120, 9341–9349. [Google Scholar] [CrossRef]
- Endres, F.; Borisenko, N.; El Abedin, S.Z.; Hayes, R.; Atkin, R. The interface ionic liquid(s)/electrode(s): In situSTM and AFM measurements. Faraday Discuss. 2012, 154, 221–233. [Google Scholar] [CrossRef]
- Carstens, T.; Lahiri, A.; Borisenko, N.; Endres, F. [Py1,4]FSI-NaFSI-Based Ionic Liquid Electrolyte for Sodium Batteries: Na+ Solvation and Interfacial Nanostructure on Au(111). J. Phys. Chem. C 2016, 120, 14736–14741. [Google Scholar] [CrossRef]
- Begić, S.; Li, H.; Atkin, R.; Hollenkamp, A.F.; Howlett, P.C. A comparative AFM study of the interfacial nanostructure in imidazolium or pyrrolidinium ionic liquid electrolytes for zinc electrochemical systems. Phys. Chem. Chem. Phys. 2016, 18, 29337–29347. [Google Scholar] [CrossRef]
- Borisenko, N.; Lahiri, A.; Pulletikurthi, G.; Cui, T.; Carstens, T.; Zahlbach, J.; Atkin, R.; Endres, F. The Au(111)/IL interfacial nanostructure in the presence of precursors and its influence on the electrodeposition process. Faraday Discuss. 2018, 206, 459–473. [Google Scholar] [CrossRef]
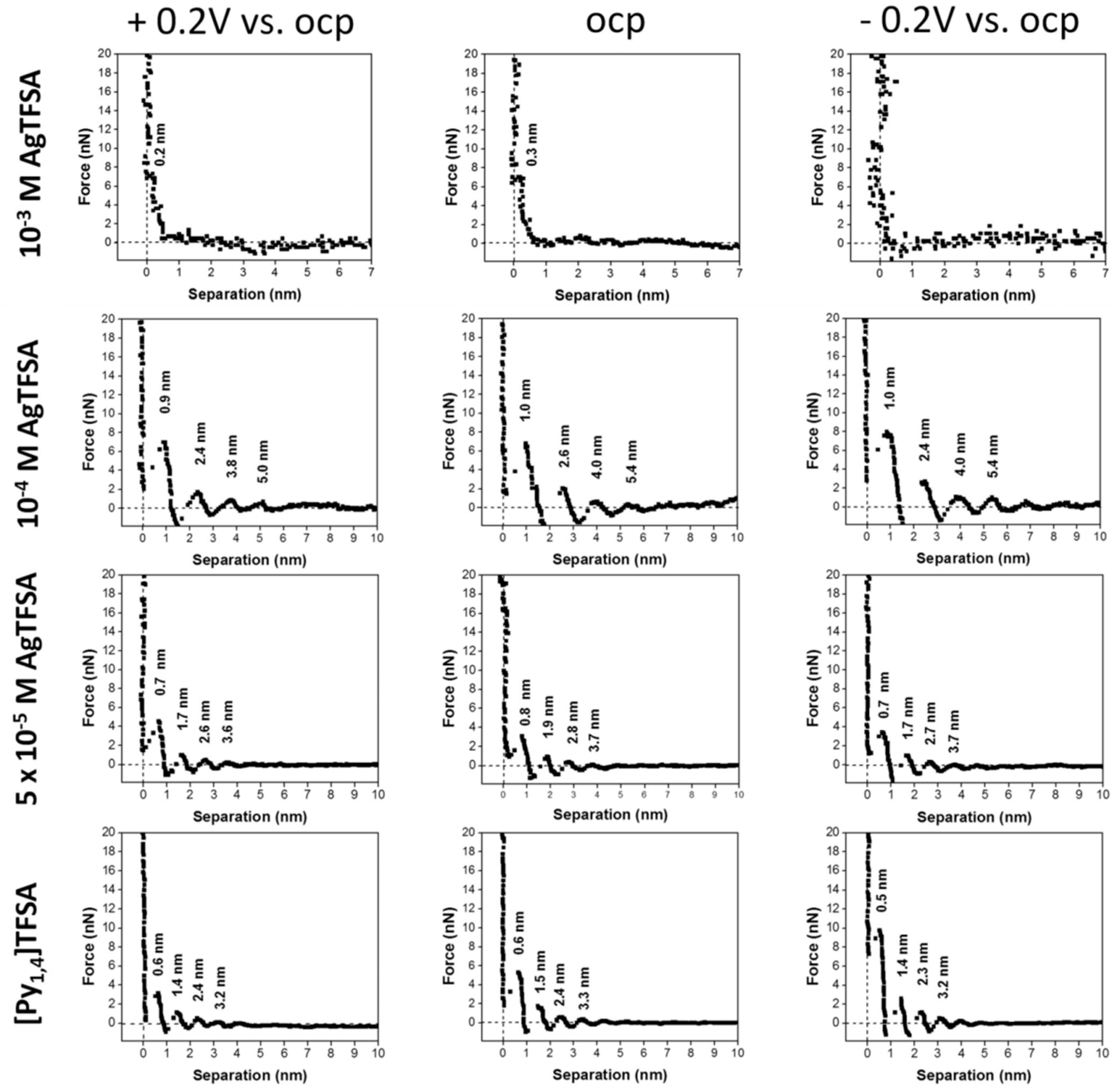
- Hoffmann, V.; Pulletikurthi, G.; Carstens, T.; Lahiri, A.; Borodin, A.; Schammer, M.; Horstmann, B.; Latz, A.; Endres, F. Influence of a silver salt on the nanostructure of a Au(111)/ionic liquid interface: An atomic force microscopy study and theoretical concepts. Phys. Chem. Chem. Phys. 2018, 20, 4760–4771. [Google Scholar] [CrossRef]
- Fukuma, T.; Kobayashi, K.; Matsushige, K.; Yamada, H. True atomic resolution in liquid by frequency-modulation atomic force microscopy. Appl. Phys. Lett. 2005, 87, 034101. [Google Scholar] [CrossRef] [Green Version]
- Siretanu, I.; Ebeling, D.; Andersson, M.P.; Stipp, S.L.S.; Philipse, A.; Stuart, M.C.; van den Ende, D.; Mugele, F. Direct observation of ionic structure at solid-liquid interfaces: A deep look into the Stern Layer. Sci. Rep. 2015, 4, 4956. [Google Scholar] [CrossRef] [PubMed]
- Black, J.M.; Baris Okatan, M.; Feng, G.; Cummings, P.T.; Kalinin, S.V.; Balke, N. Topological defects in electric double layers of ionic liquids at carbon interfaces. Nano Energy 2015, 15, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Jain, S. Shilpa Defects and Order in Liquid Crystal Phases. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 1999. [Google Scholar]
- Negami, M.; Ichii, T.; Murase, K.; Sugimura, H. Visualization of Ionic-Liquid/Solid Interfaces by Frequency Modulation Atomic Force Microscopy. ECS Trans. 2013, 50, 349–355. [Google Scholar] [CrossRef]
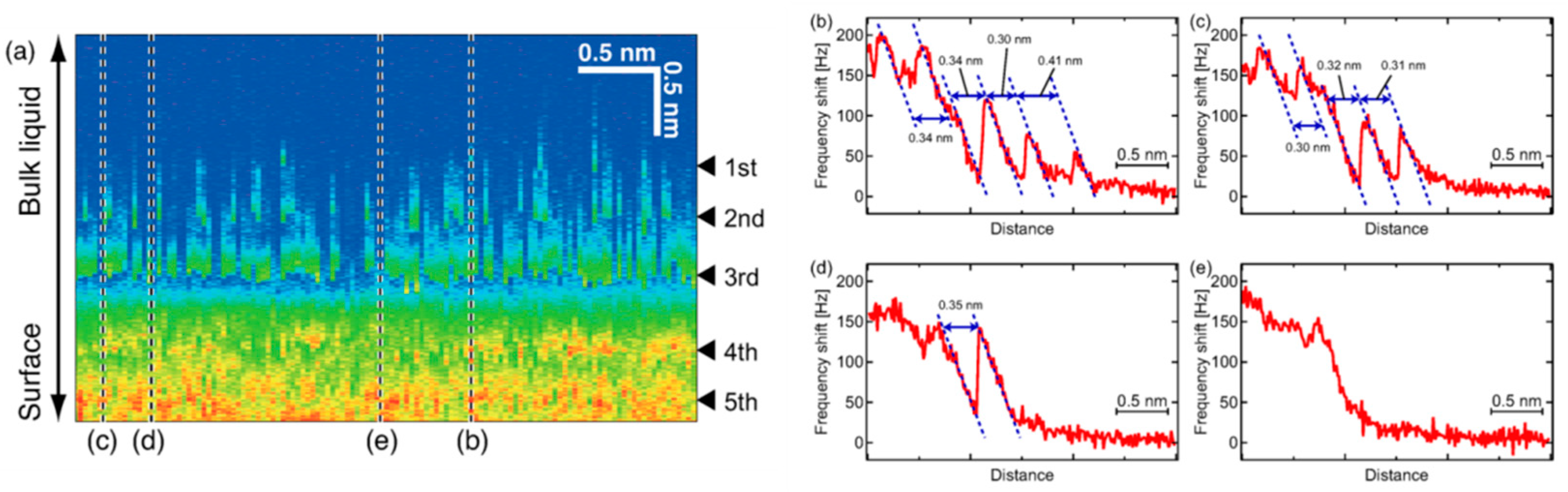
- Ichii, T.; Negami, M.; Sugimura, H. Atomic-Resolution Imaging on Alkali Halide Surfaces in Viscous Ionic Liquid Using Frequency Modulation Atomic Force Microscopy. J. Phys. Chem. C 2014, 118, 26803–26807. [Google Scholar] [CrossRef]
- Page, A.J.; Elbourne, A.; Stefanovic, R.; Addicoat, M.A.; Warr, G.G.; Voïtchovsky, K.; Atkin, R. 3-Dimensional atomic scale structure of the ionic liquid–graphite interface elucidated by AM-AFM and quantum chemical simulations. Nanoscale 2014, 6, 8100–8106. [Google Scholar] [CrossRef]
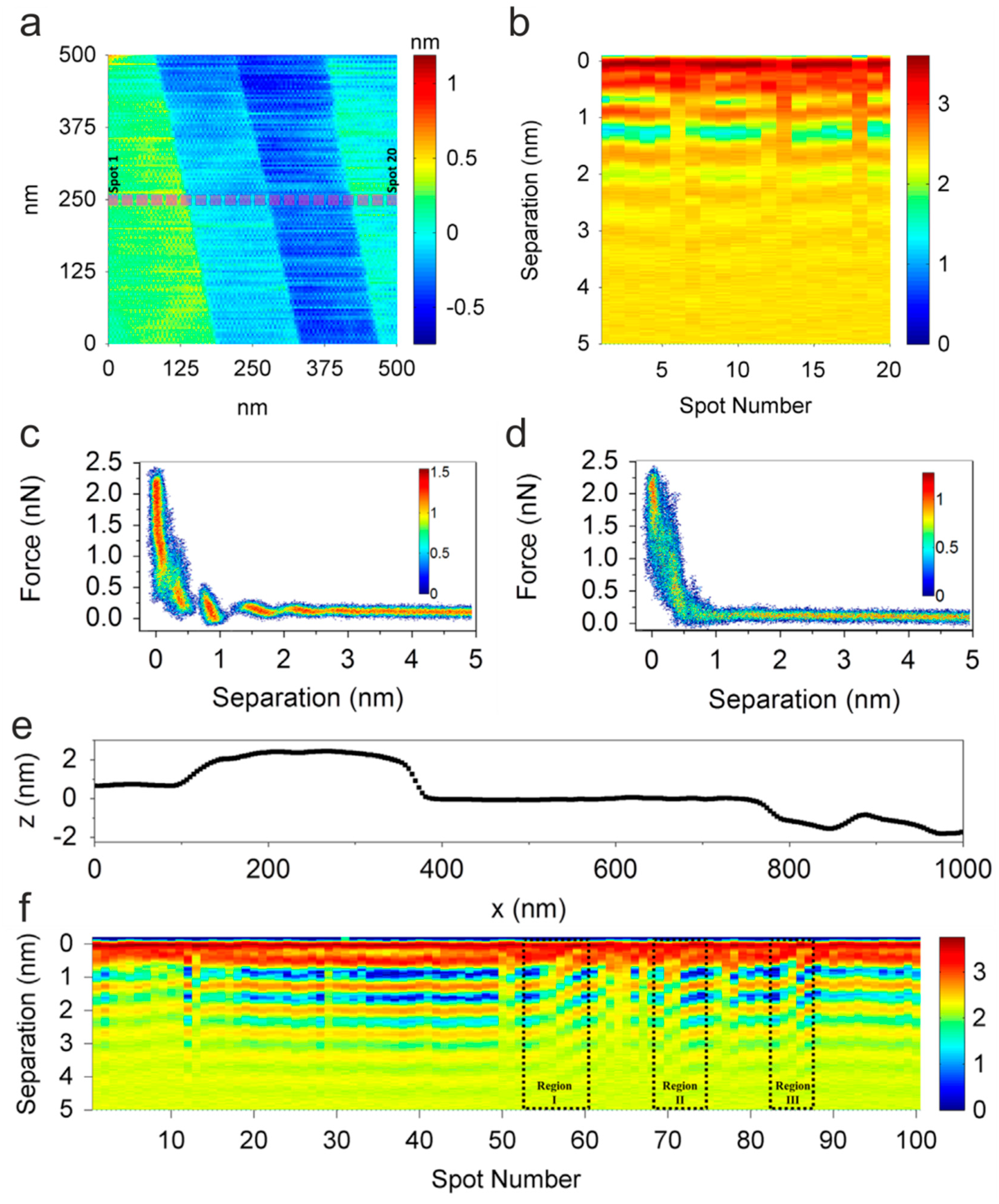
- Elbourne, A.; McDonald, S.; Voïchovsky, K.; Endres, F.; Warr, G.G.; Atkin, R. Nanostructure of the Ionic Liquid–Graphite Stern Layer. ACS Nano 2015, 9, 7608–7620. [Google Scholar] [CrossRef]
- Ebeling, D.; Bradler, S.; Roling, B.; Schirmeisen, A. 3-Dimensional Structure of a Prototypical Ionic Liquid–Solid Interface: Ionic Crystal-Like Behavior Induced by Molecule–Substrate Interactions. J. Phys. Chem. C 2016, 120, 11947–11955. [Google Scholar] [CrossRef]



















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodenbücher, C.; Wippermann, K.; Korte, C. Atomic Force Spectroscopy on Ionic Liquids. Appl. Sci. 2019, 9, 2207. https://doi.org/10.3390/app9112207
Rodenbücher C, Wippermann K, Korte C. Atomic Force Spectroscopy on Ionic Liquids. Applied Sciences. 2019; 9(11):2207. https://doi.org/10.3390/app9112207
Chicago/Turabian StyleRodenbücher, Christian, Klaus Wippermann, and Carsten Korte. 2019. "Atomic Force Spectroscopy on Ionic Liquids" Applied Sciences 9, no. 11: 2207. https://doi.org/10.3390/app9112207
APA StyleRodenbücher, C., Wippermann, K., & Korte, C. (2019). Atomic Force Spectroscopy on Ionic Liquids. Applied Sciences, 9(11), 2207. https://doi.org/10.3390/app9112207