1. Introduction
Broadband pulse light sources, such as a Ti:Sapphire laser and other femtosecond (fs) laser systems, enable a range of important applications, such as nonlinear and time-resolved spectroscopic studies of molecular dynamics and chemical reactions [
1,
2,
3,
4], coherent anti-Stokes Raman scattering spectroscopy [
5], and even laser annealing [
6]. In addition, ultrashort optical pulses in the deep ultraviolet (DUV) region can be used for the resonant and non-resonant multi-photon ionization of molecules in mass spectrometry, which can be applied to improve reliability in forensic science [
7,
8]. However, for studying fast processes in atoms and molecules, such as electron movement [
9], one needs a source of coherent radiation producing pulses on the order of several fs or even beyond that [
10].
Production of ever-shorter light pulses requires generation of an emission with an ever-wider coherent bandwidth and careful control of the phases. One of the methods for generating ultrabroadband spectrum is termed molecular modulation [
11,
12,
13]. Several demonstrations of the ultrabroadband light source have been reported, i.e., the generation of more than 40 emission lines with a picosecond laser in hydrogen gas [
14] and more than one-octave spanning spectrum of carrier-envelope controlled Raman sidebands [
15]. Such multi-emissions can be employed not only as an ultrabroadband laser system, but also as a source of ultrashort optical pulses if the phases of the multiple molecular modulation sidebands can be properly controlled. Manipulation of the relative phases is typically possible with a spatial light modulator (SLM) [
16] or a spatial phase controller [
17]. Then, the resultant temporal waveform can be measured by, for example, cross-correlation signals in Xe gas [
18].
In this work, we used a picosecond laser system to demonstrate molecular modulation in ambient air. We utilized a collinearly focused pump and Stokes beams with the optical frequency difference matching a vibrational transition of nitrogen molecules. In a relatively simple experimental configuration we produced a broad spectrum of multiple mutually-coherent narrow-band components, and demonstrated control over the spectral phases of these components. The adjustment of the spectral phases was done without separating the spectral components spatially, but rather by adjusting an overall dispersion via varying the optical path in glass. Furthermore, the diagnostics of the resultant pulsed waveform was performed by refocusing the multi-component beam into the second interaction region and utilizing molecular modulation in air the second time.
In the following we describe the procedure for generating multiple coherent Raman sidebands in air using the vibrational mode of the nitrogen molecule at 2331 cm−1. We then demonstrate how the relative phases of the sidebands can be controlled by a pair of rotating glass plates. Finally, we determine the phases of the Raman sidebands and estimate the temporal waveform of the generated ultrashort pulses by monitoring the optical spectrum of the third anti-Stokes sideband (AS3) with a spectrometer.
2. Experimental Setup
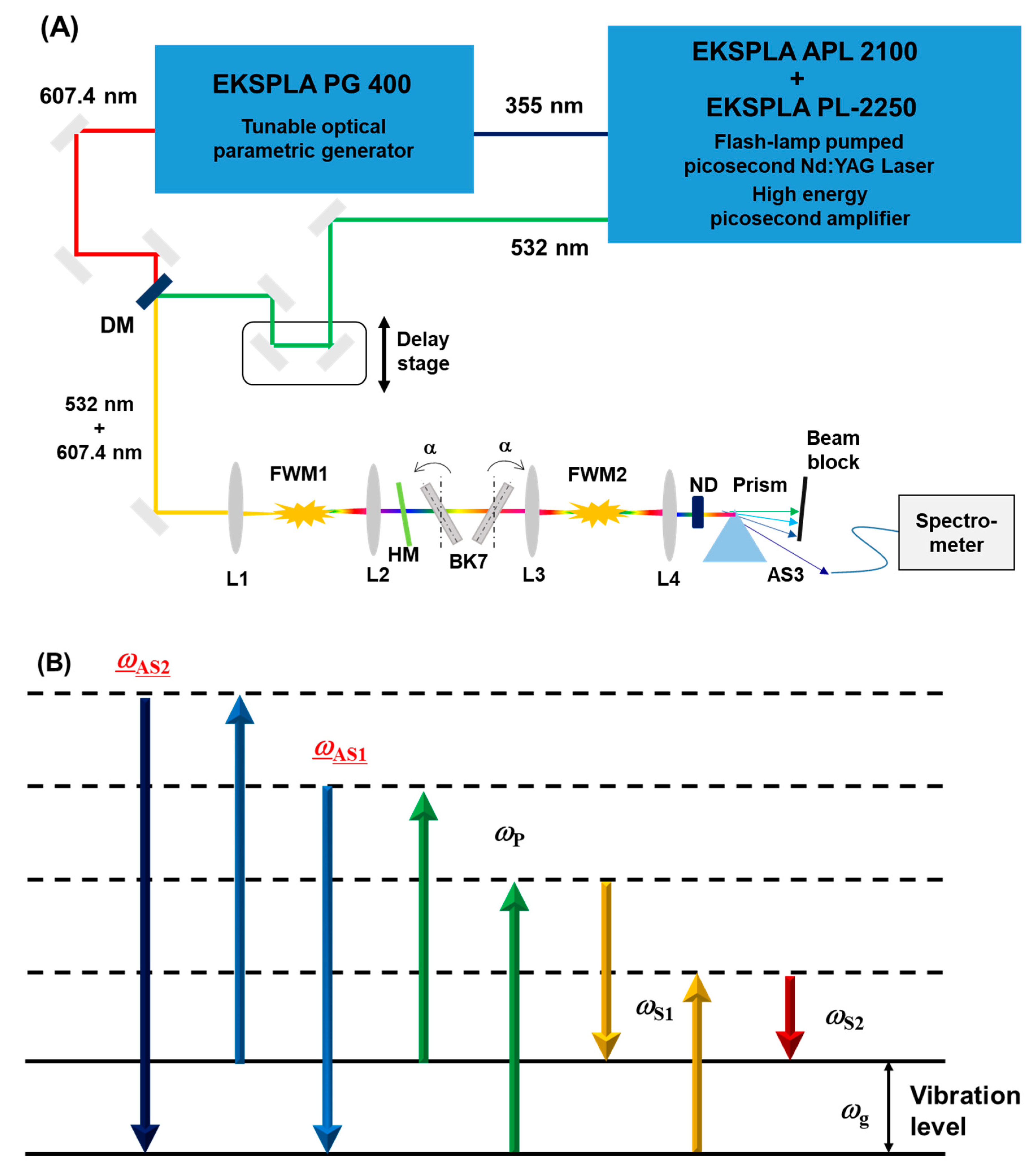
Figure 1 shows the experimental setup. We employed a flash-lamp pumped picosecond (ps) Nd:YAG laser (APL2100, EKSPLA) and a regenerative amplifier system (PL-2250, EKSPLA). The laser system produces up to 400 mJ 1064 nm Fourier transform limited pulses (~50 ps full width at half maximum (FWHM)) at 10 Hz repetition rate. The laser system has optional outputs with second (532 nm) and third (355 nm) harmonics of the fundamental radiation. For the following experiments we used the second harmonic (SH) pulse, with the average energy 20.6 mJ and less than 1% pulse-to-pulse energy fluctuations. The third harmonic (TH), in its turn, pumps the tunable optical parametric oscillator (OPG) system (PG 400, EKSPLA). The OPG produces 607.4 nm pulses with the average energy 2.6 mJ (6% fluctuations). We chose the operating wavelength of the OPG so that the frequency difference between the generated pulse (Stokes) and 532 nm (pump) pulse matches the fundamental vibrational frequency of nitrogen molecule (2331 cm
−1). The pump pulse is appropriately time delayed for maximizing temporal overlap with the 607.4 nm pulse and then spatially combined with it by a dichroic mirror (DM). Afterwards, the collinearly propagating beams are focused in the air (FWM1,
Figure 1) by the convex lens L1 (f = 150 mm) and then collimated by the lens L2 (f = 100 mm). The two laser pulses overlap temporally and spatially in the focal plane of the lens, where they generate multiple Raman sidebands. The energy level of this process is shown in
Figure 1B. The frequency difference between the adjacent sidebands equals the fundamental vibrational frequency by nitrogen molecule. We note that chromatic aberrations of the focusing would have provided only a limited detrimental effect on the sidebands generation as all the pump, Stokes, and generated sidebands propagate collinearly, and the actual interaction region is on the order of the Rayleigh length in our experiment (0.5 mm) [
19]. In the experiment, we confirmed the ideal spatial and temporal overlaps by maximizing the generated sidebands energies after FWM1 point.
Before the beams interacted the second time (FWM2,
Figure 1), we equalized the energies of the pump, Stokes, and the generated sidebands by properly attenuating the pump and Stokes pulses with the 532 nm high reflection mirror (HM,
Figure 1). The HM mirror reflects more than 95% of the pump and 50% of Stokes radiation, so the intensity of the Stokes pulse becomes approximately the same as the intensity of the first generated anti-Stokes sideband. After the HM, the attenuated pump, Stokes pulses, and the sidebands pass through a pair of 12-mm thickness N-BK7 glass plates. These plates are installed in mirror mounts equipped with precise actuators (Newport, mirror mount: U200-A3K, actuator: AJS100-0.5K-NL). This allows us to accurately control the incident angle of the input beams, which results in change of the optical path through the glass (
Figure S1 in the Supplementary Materials). The change of the optical path, in turns, lead to the modification of the relative phase among several pulses [
20]. By using two glass plates and rotating them in opposite direction, we can change the optical paths of the pulses without shifting the directions of the beams.
First, we set two mirror mounts with BK7 glasses at an angle of 27 degrees (written
α in
Figure 1). After passing through these two BK7 plates, the multiple sidebands were focused in the air again (FWM2,
Figure 1). The 3rd anti-Stokes (AS3 at 387 nm) generated at FWM2 was separated from others with the prism, and its spectrum recorded with a fiber-coupled multichannel spectrometer (HR 4000, Ocean Optics, integration time: 150 msec, average: 100 times). We used this configuration to record the peak signal of AS3 sideband while changing the angle of the glass plates. We performed these measurements for the angle of the glass plates from 27.00 to 32.09 degrees.
3. Results and Discussions
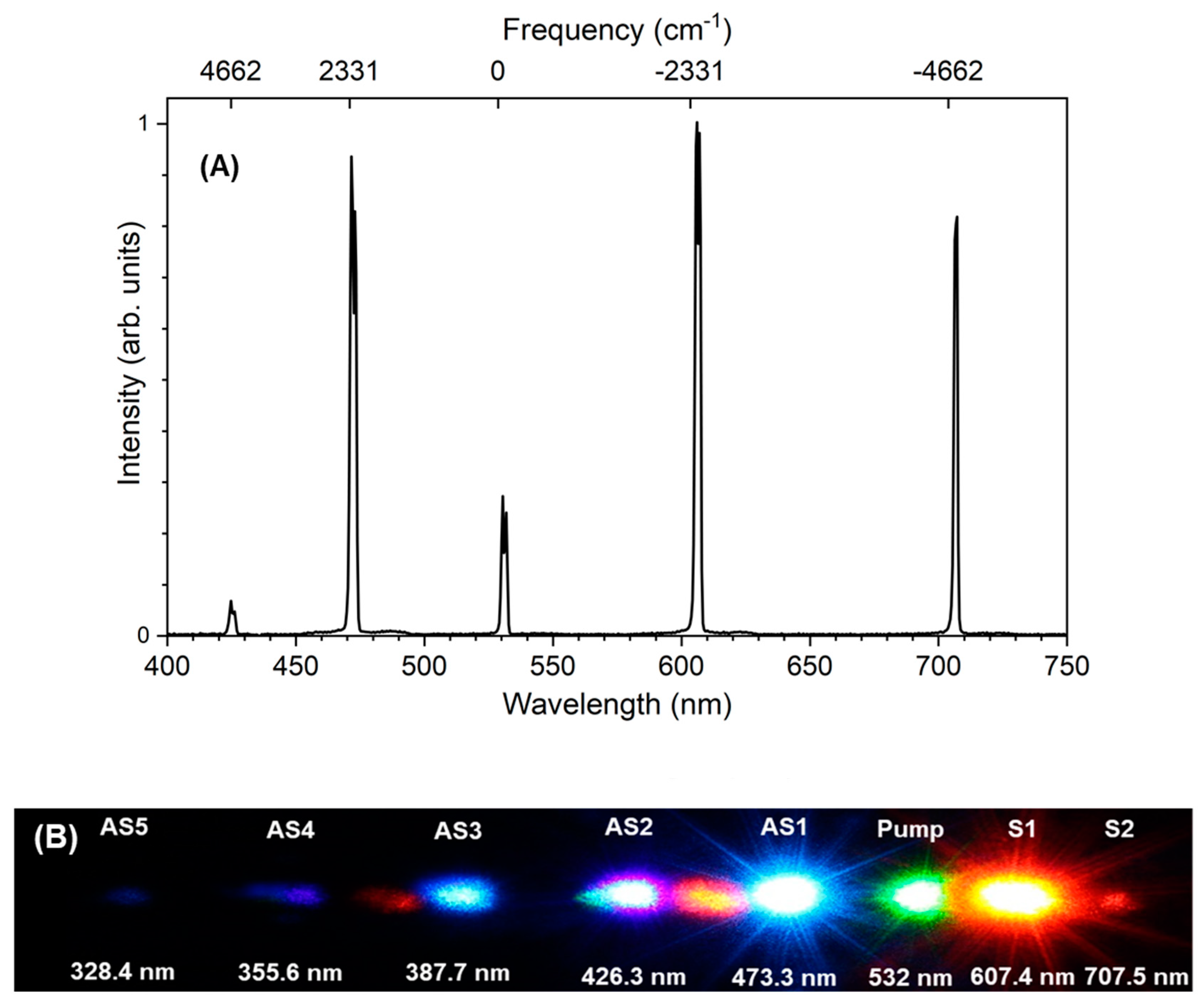
Figure 2 shows the spectrum obtained after the 532 nm high reflection mirror. We observed several sidebands, from the second order anti-Stokes (AS2, 426 nm) to the second order Stokes (S2, 707 nm). Due to a high reflectivity at 532 nm for the mirror (HM), the intensity of the pump pulse was smaller than the first order anti-Stokes (AS1) and Stokes (S1) pulses. The upper axis shows the frequency shift from the pump beam. We see that the frequency spacing of the sidebands are in reasonable agreement with the nitrogen vibrational frequency (2331 cm
−1), despite the small shift due to the spectrometer calibration. Hence, we conclude that the sidebands are generated via molecular modulation of nitrogen molecules.
In what follows, we describe an experiment where the sidebands produced in the first focal region (FWM1,
Figure 1) are refocused into the second region (FWM2,
Figure 1) where we use their coherent interaction with nitrogen molecules for diagnostics of the resultant optical waveform. The refocused sidebands produced molecular coherence which is not only a function of their intensities but also their relative phases. Then, additional higher-order sidebands were produced in proportion to the magnitude of this molecular excitation. We controlled the relative phases of the primary sidebands by varying the amount of dispersion due to tilted BK7 glass plates in the beam path.
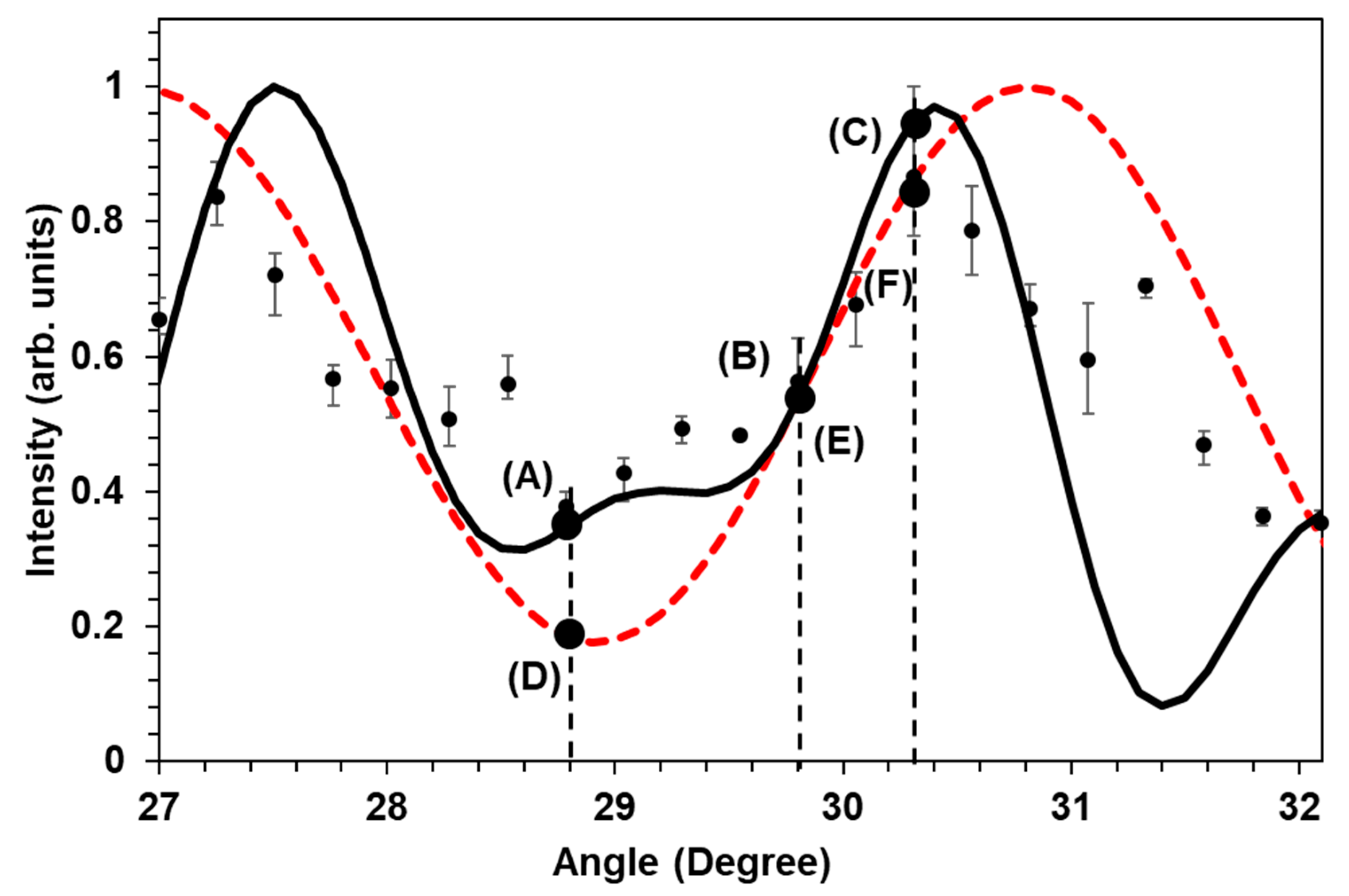
Figure 3 shows the intensity modulation of the AS3 sideband at 386.87 nm after the FWM2 point, at various tilt angles of the BK7 plates. The relative phase among the sidebands changed because of the different optical path lengths and the dispersion. When the sidebands are mutually coherent they interfere with each other and produce short pulses.
We assume that the driving force producing the molecular coherence is proportional to the absolute value of the electric field amplitude, squared. This assumption is justified when the resultant coherence
is relatively small (
, which is the case under our experimental conditions), or when the molecular excitation is adiabatic [
14]. The vibrational coherence, in turn, mixes with the highest-order anti-Stokes (AS2) sideband (which at this point is presumed to be of constant intensity) to produce the next anti-Stokes (AS3) sideband [
21]. In this case, the AS3 signal can be evaluated by the equation:
where
are constants from the interaction Hamiltonian for molecular nitrogen [
22], and
is the slowly-varying complex electric field amplitude at frequency
corresponding to the
th-order (pump, Stokes, or anti-Stokes) sideband, such that the total electric field is:
To properly evaluate phase
for each sideband we need to determine the optical path length, which depends on the wavelength of each sideband. Hence, we used the Sellmeier equation to estimate the refractive index and apply Snell’s law to calculate refraction angles and the optical path length for each individual sideband
[
23,
24]. The exact procedure is described in the
Supplementary Materials. Then we used the obtained sideband phase in conjunction with Equation (1) to plot the theoretical curves of
Figure 3. We took the power ratio between the sidebands AS2:AS1:P:S1:S2 as 0.05:1:0.3:1:0.8, corresponding to the experimentally measured spectra and assumed time-constant sideband intensities, essentially limiting our analysis to a time window of a few-picosecond centered around the peak of the applied pump/Stokes pulse envelope, and assuming that the total envelope duration was much longer than that. In case of 5-sidebands contributing to generation at the second focus (FWM2), Equation (1) can be written as:
Best fit to the data shown in
Figure 3 gives the initial relative phases among the five sidebands as: AS2—4.4 rad, AS1—0 rad, P—0 rad, S1—1.8 rad, S2—5.4 rad. Note that among the five sideband phases only three are independent, and two can be made zero, as long as the total waveform duration is much longer than the optical period and carrier-envelope phase effects can be neglected. In addition, we checked what is expected in the case of only three sidebands contributing to the molecular excitation in the second focal region (FWM2,
Figure 1). In that case, Equation (1) becomes:
Here, by the best-fit procedure, we extract the following initial phases: AS2—4.3 rad, AS1—0 rad, P—0 rad. The results of the two theoretical calculations, along with the experimental measurements, are shown in
Figure 3 (solid and dashed curves). As we can see from
Figure 3, both theoretical curves generally match the experimental results, however, there are discrepancies at several experimental points.
One of the possible explanations for the mismatch between the experimental and theoretical data is the non-perfect phase-matching in the Raman generation process in the second focal region. In the above-described theory, we assume the ideal phase-matching case, i.e., the zero wave vector mismatch for the included sidebands. Even in air where the dispersion in the visible, near-ultraviolet and, infrared spectral regions is small, it cannot be completely neglected, particularly in the case of broadband multi-sideband generation. The experimentally measured modulation of the AS3 signal with the change of the optical path suggests that we can partially compensate for the dispersion of air by the rotating pair of glass plates. Moreover, the modulation confirms the coherent nature of the sidebands and suggests a way, by the manipulation of the relative phases among the sidebands, to control the waveform of the formed pulse train. Essentially, variable dispersion due to the tilted glass plates affords sufficient phase control, when the number of sidebands is not too large. In principle, more sideband phases can be more precisely controlled if several different types of glass are used [
17,
25].
To determine the intensity envelope of the pulse-train waveform (which corresponds to the superposition of sideband fields) we used the following equation:
Here
are the initial sideband phases obtained from the best fit procedure described above. The more explicit notation
emphasizes the fact that the individual sideband phases vary with the varying glass-plate tilt angle
. Each phase here is a different function of
, due to glass dispersion.
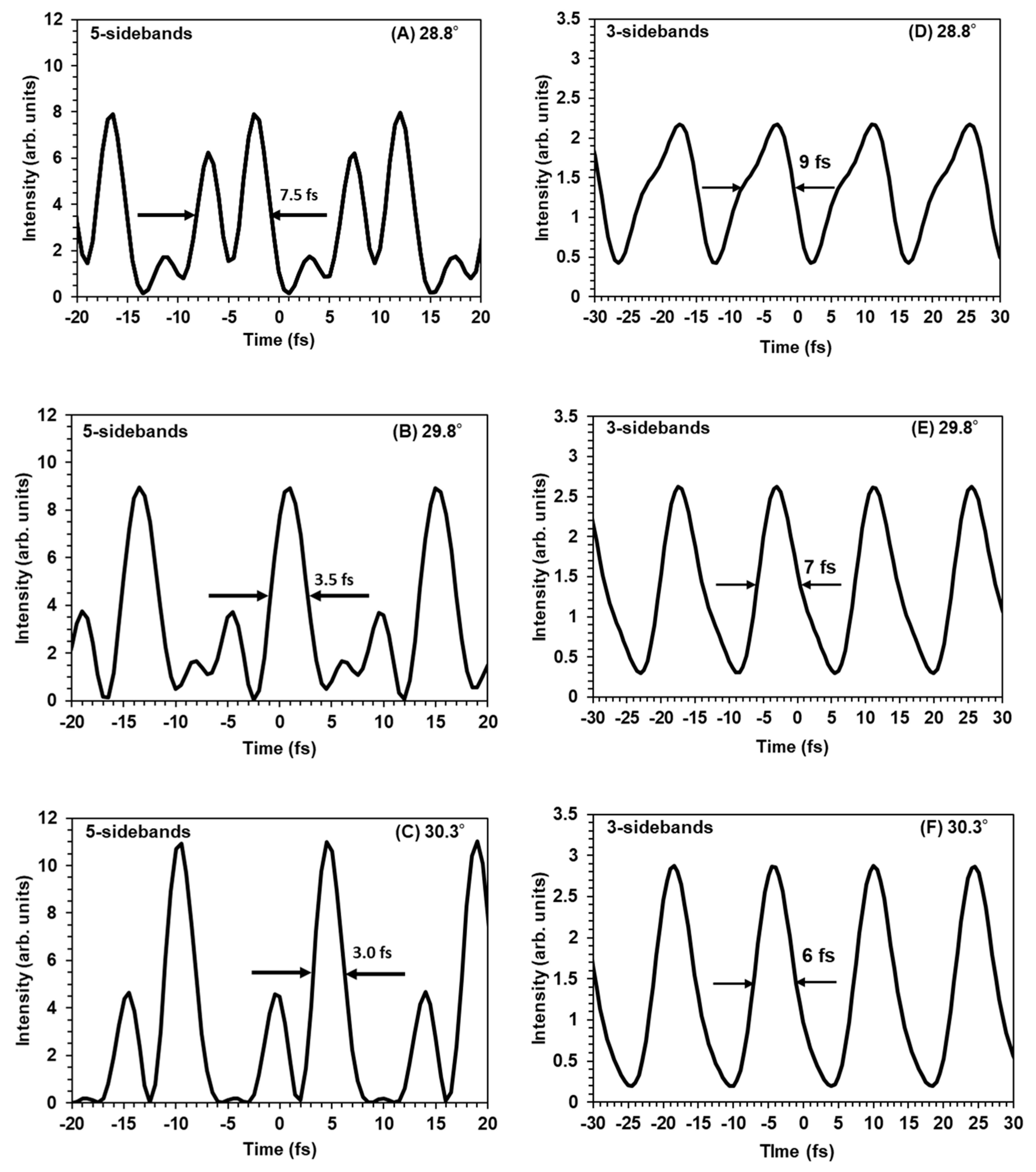
Figure 4 shows the predicted waveforms obtained for various points on the AS3 modulation curve in
Figure 3.
We calculated the temporal waveforms for six points: (A), (B), (C), (D), (E) and (F), shown in
Figure 3. Each of these points corresponds to a different incident angle on the rotating BK7 glass. As we can see, in the case of the 5-sidebands contribution (
Figure 4A–C) the overall change of the pulse train lead to shortening of each individual pulse: from 7.5 fs to 3 fs FWHM duration at the optimum angle (30.3°). On the other hand, for the case of 3-sidebands contribution (
Figure 4D–F), we notice that the pulse had changed from 9 fs to 6 fs at the same optimum angle of 30.3°. As the spectra have more components and as phase compensation with the glass dispersion simultaneously for the 5 sidebands is more difficult than for the 3 sidebands case, the 5-sidebands simulated waveform has a more complicated structure with sub-pulses. If these sub-pulses become larger, then the train of single fs pulses will transform into the pulse train of double pulses, as we can see in
Figure 4A, which makes the effective pulse duration measured at FWHM twice larger.
However, for both the cases the peak intensity of the simulated temporal waveform increased as the AS3 signal became higher. These results suggest that the multiple sidebands are mutually coherent in the picosecond regime, generating a fs pulse train. Hence, by measuring one of the anti-Stokes signals we retrieved the temporal waveform synthesized by multiple sidebands.




{kind=link}
{kind=link}
{kind=link}
{kind=link}



