Light Harvesting and Optical-Electronic Properties of Two Quercitin and Rutin Natural Dyes


Abstract
:1. Introduction
2. Experiment and Theory
2.1. Experiment
2.2. Theory
3. Results and Discussion
3.1. Geometric Structures
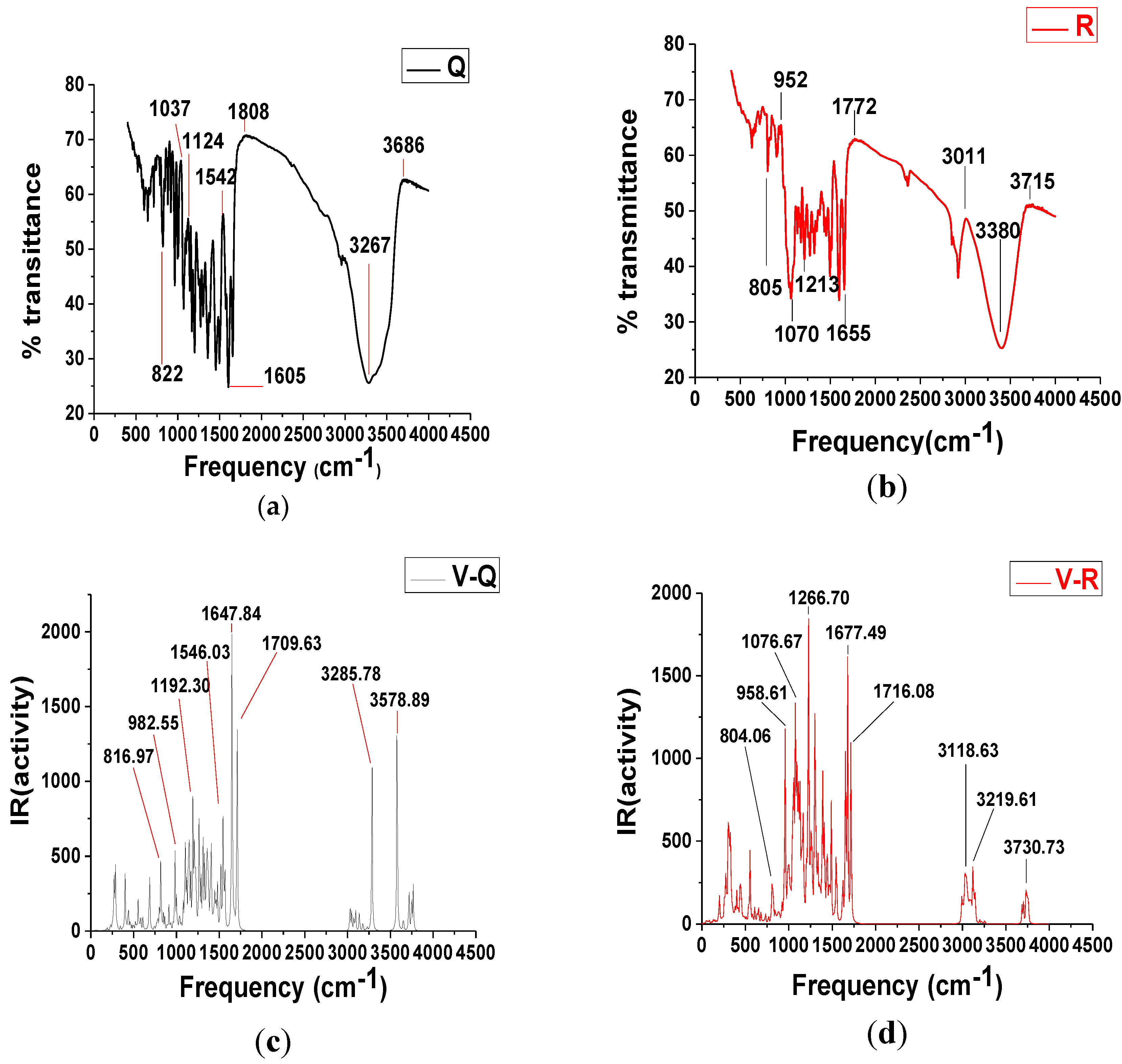
3.2. Fourier Transforms Infrared Spectrum
3.3. Projected Density of State Analysis
3.4. NBO Analysis
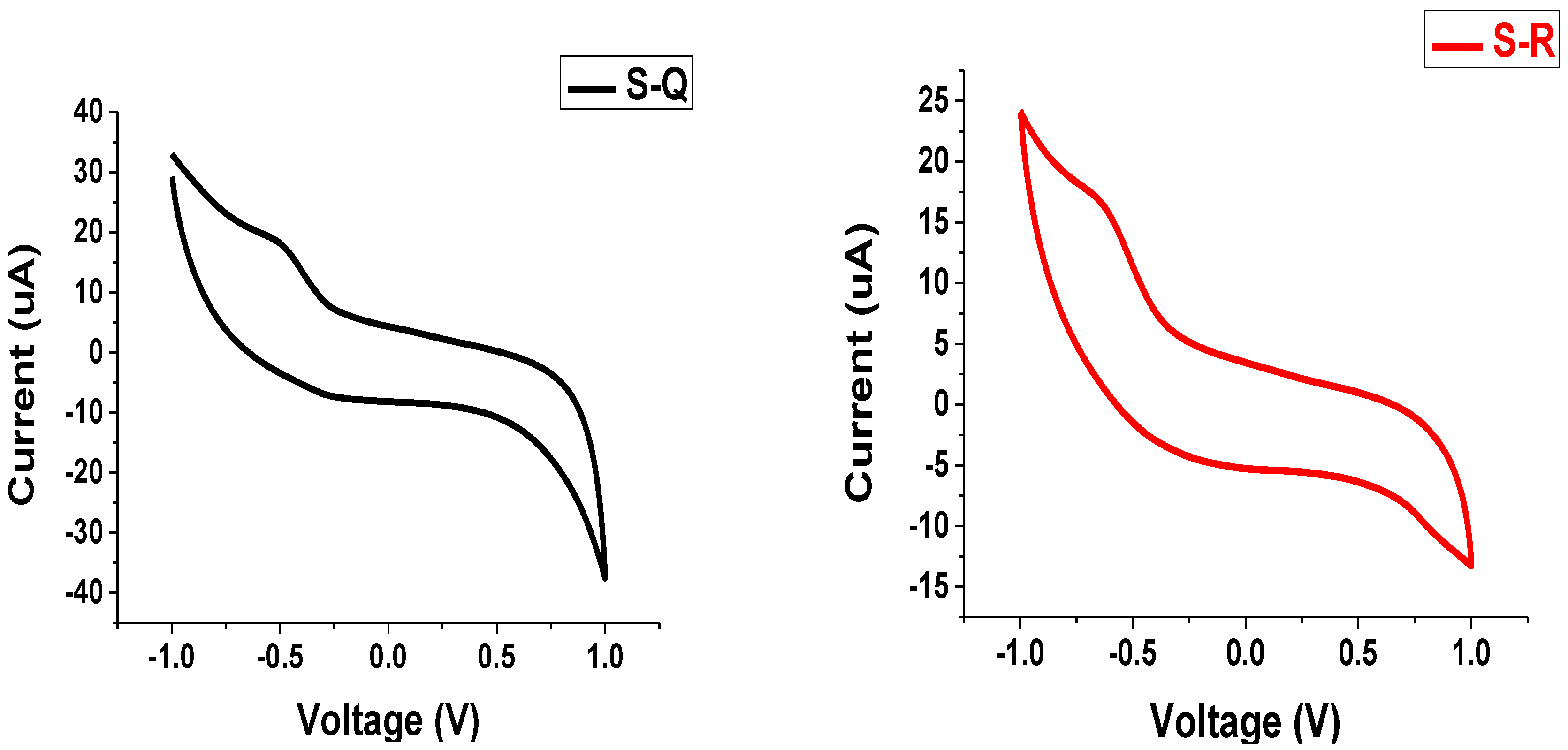
3.5. Electrochemical Characteristics
3.6. The Molecular Descriptor Parameters Analysis
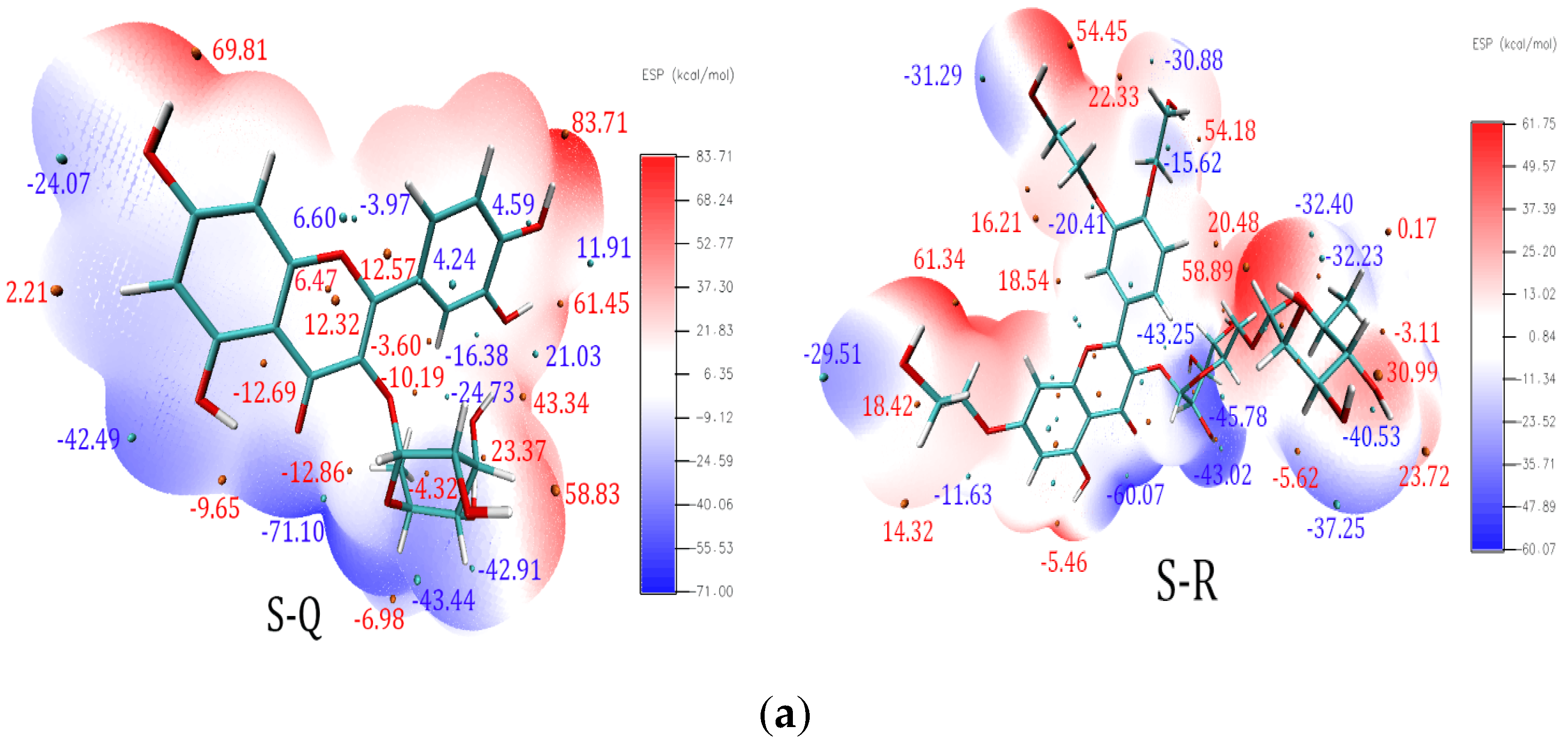
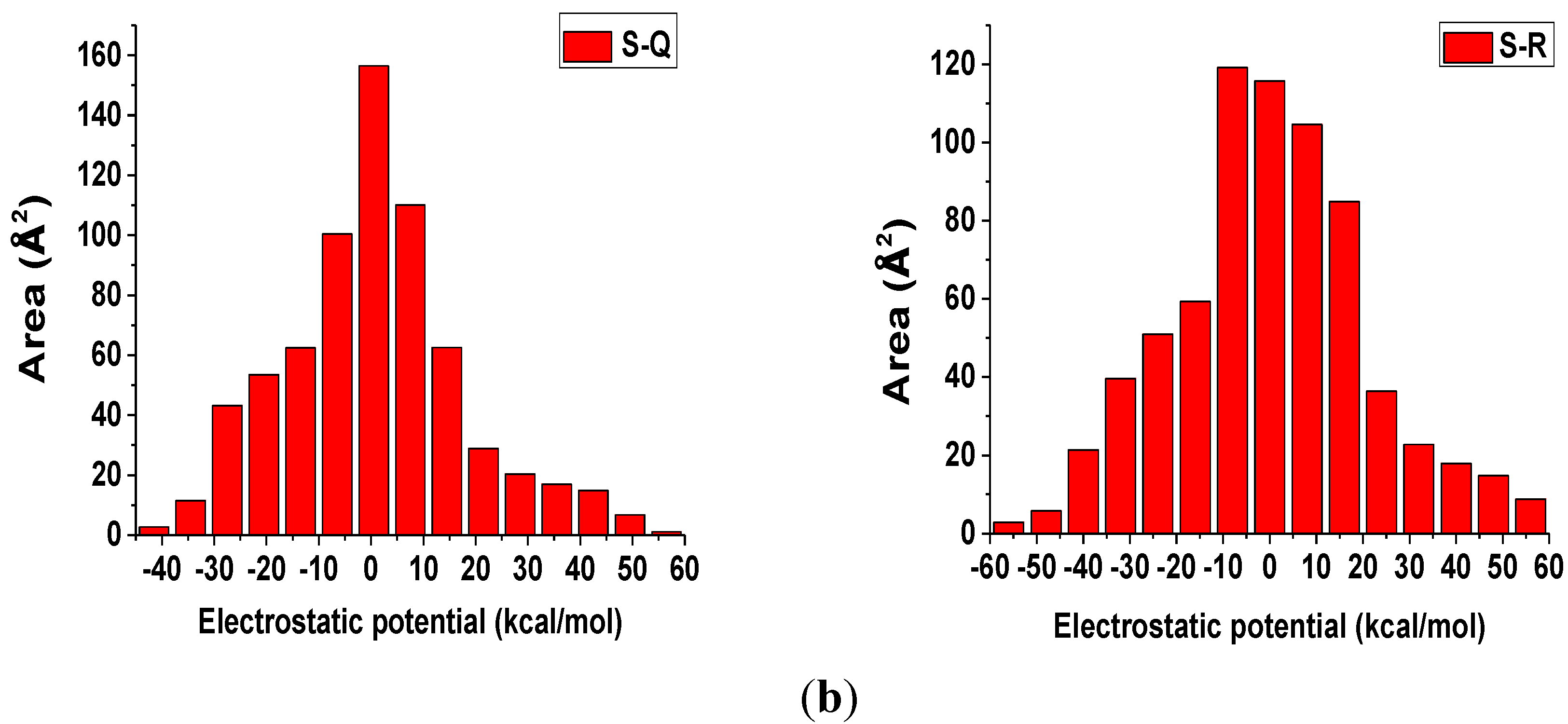
3.7. ESP Analysis
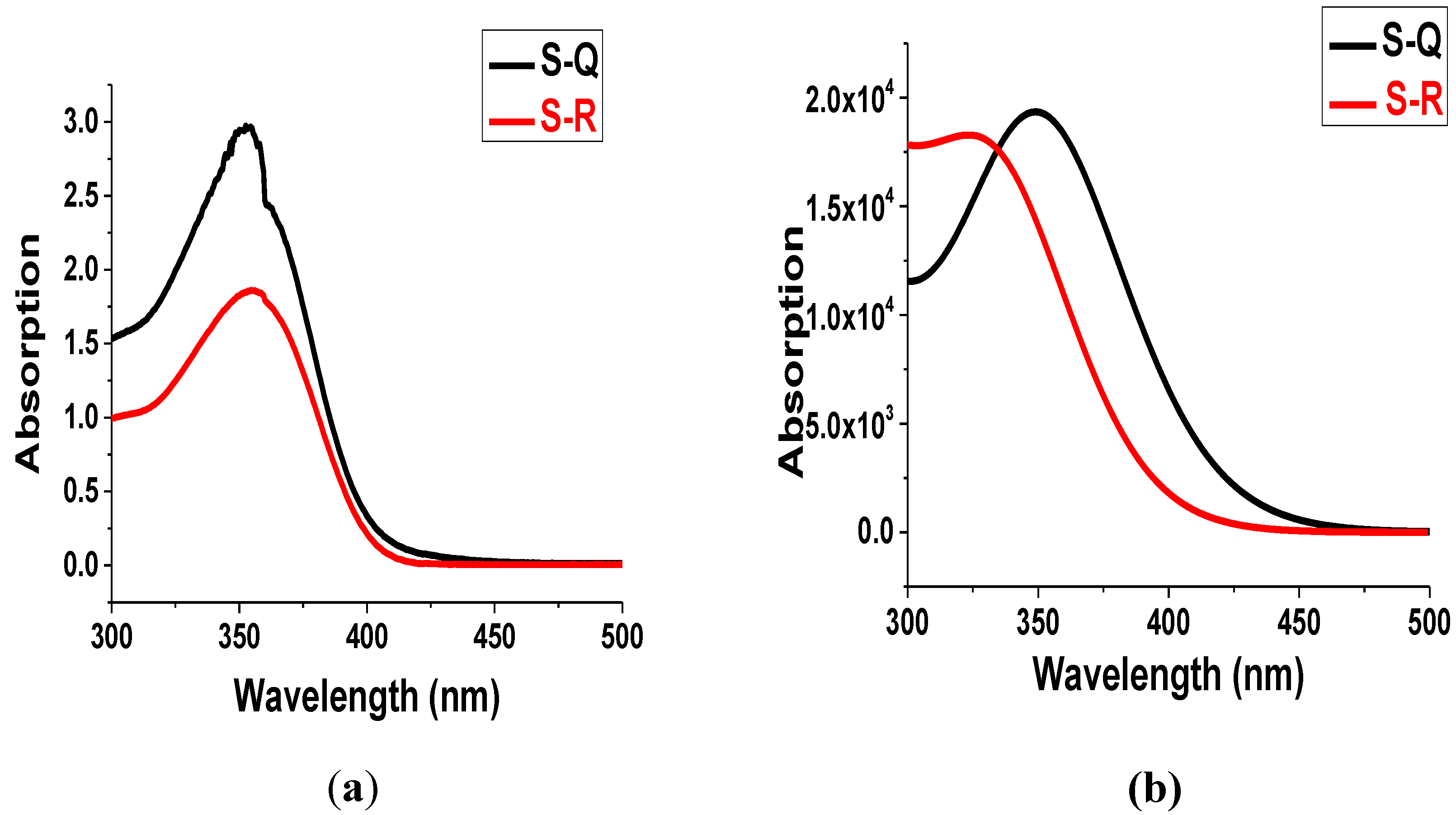
3.8. Absorption Properties
3.9. Polarizability, First Hyperpolarizability and Charge Transfer Performance
3.10. Reorganization Energies
3.11. The Fluorescent Lifetime of S-Q and S-R
3.12. Photovoltaic Characteristics of S-Q and S-R
3.13. Prediction for Photovoltaic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, H.L.; Dai, H.J. Strongly coupled inorganic-nano-carbon hybrid materials for energy storage. Chem. Soc. Rev. 2013, 42, 3088–3113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Ye, D.J. Forecasting Chinese carbon emissions from fossil energy consumption using non-linear grey multivariable models. J. Clean. Prod. 2017, 142, 600–612. [Google Scholar] [CrossRef]
- Baul, T.K.; Datta, D.; Alam, A. A comparative study on household level energy consumption and related emissions from renewable (biomass) and non-renewable energy sources in Bangladesh. Energy Policy. 2018, 114, 598–608. [Google Scholar] [CrossRef]
- Wang, J.G.; Mu, X.J.; Wang, X.X.; Wang, N.; Ma, F.C.; Liang, W.J.; Sun, M.T. The thermal and thermoelectric properties of in-plane C-BN hybrid structures and graphene/h-BN van der Waals heterostructures. Mater. Today Phys. 2018, 5, 29–57. [Google Scholar] [CrossRef]
- Pandey, A.K.; Hossain, M.S.; Tyagi, V.V.; Abd Rahim, N.; Selvaraj, J.A.L.; Sari, A. Novel approaches and recent developments on potential applications of phase change materials in solar energy. Renew. Sustain. Energy Rev. 2018, 82, 281–323. [Google Scholar] [CrossRef]
- Breyer, C.; Bogdanov, D.; Aghahosseini, A.; Gulagi, A.; Child, M.; Oyewo, A.S.; Farfan, J.; Sadovskaia, K.; Vainikka, P. Solar photovoltaics demand for the global energy transition in the power sector. Prog. Photovolt. 2018, 26, 505–523. [Google Scholar] [CrossRef]
- Ali, B.A.; Allam, N.K. Propping the optical and electronic properties of potential photo-sensitizers with different π-spacers: TD-DFT insights. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 188, 237–243. [Google Scholar] [CrossRef]
- Peksu, E.; Karaagac, H. A third generation solar cell based on wet-chemically etched Si nanowires and sol-gel derived Cu2ZnSnS4 thin films. J. Alloy. Compd. 2019, 774, 1117–1122. [Google Scholar] [CrossRef]
- Nozik, A.J.; Beard, M.C.; Luther, J.M.; Law, M.; Ellingson, R.J.; Johnson, J.C. Semiconductor Quantum Dots and Quantum Dot Arrays and Applications of Multiple Exciton Generation to Third-Generation Photovoltaic Solar Cells. Chem. Rev. 2010, 110, 6873–6890. [Google Scholar] [CrossRef]
- Han, L.; Liu, J.; Liu, Y.; Cui, Y.H. Novel D-A-pi-A type benzocarbazole sensitizers for dye sensitized solar cells. J. Mol. Struct. 2019, 1180, 651–658. [Google Scholar] [CrossRef]
- Ye, M.D.; Wen, X.R.; Wang, M.Y.; Iocozzia, J.; Zhang, N.; Lin, C.J.; Lin, Z.Q. Recent advances in dye-sensitized solar cells: From photoanodes, sensitizers and electrolytes to counter electrodes. Mater. Today. 2015, 18, 155–162. [Google Scholar] [CrossRef]
- Sugathan, V.; John, E.; Sudhakar, K. Recent improvements in dye sensitized solar cells: A review. Renew. Sustain. Energy Rev. 2015, 52, 54–64. [Google Scholar] [CrossRef]
- Ludin, N.A.; Mahmoud, A.; Mohamad, A.B.; Kadhum, A.A.H.; Sopian, K.; Karim, N.S.A. Review on the development of natural dye photosensitizer for dye-sensitized solar cells. Renew. Sustain. Energy Rev. 2014, 31, 386–396. [Google Scholar] [CrossRef]
- Shalini, S.; Prabhu, R.B.; Prasanna, S.; Mallick, T.K.; Senthilarasu, S. Review on natural dye sensitized solar cells: Operation, materials and methods. Renew. Sustain. Energy Rev. 2015, 51, 1306–1325. [Google Scholar] [CrossRef]
- Sanjay, P.; Deepa, K.; Madhavan, J.; Senthil, S. Optical, spectral and photovoltaic characterization of natural dyes extracted from leaves of Peltophorum pterocarpum and Acalypha amentacea used as sensitizers for ZnO based dye sensitized solar cells. Opt. Mater. 2018, 83, 192–199. [Google Scholar] [CrossRef]
- Bella, F.; Gerbaldi, C.; Barolo, C.; Gratzel, M. Aqueous dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3431–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumara, N.T.R.N.; Lim, A.; Lim, C.M.; Petra, M.I.; Ekanayake, P. Recent progress and utilization of natural pigments in dye sensitized solar cells: A review. Renew. Sustain. Energy Rev. 2017, 78, 301–317. [Google Scholar] [CrossRef]
- Richhariya, G.; Kumar, A.; Tekasakul, P.; Gupta, B. Natural dyes for dye sensitized solar cell: A review. Renew. Sustain. Energy Rev. 2017, 69, 705–718. [Google Scholar] [CrossRef]
- Gong, J.; Sumathy, K.; Qiao, Q.; Zhou, Z. Review on dye-sensitized solar cells (DSSCs): Advanced techniques and research trends. Renew. Sustain. Energy Rev. 2017, 68, 234–246. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Kabir, F.; Bhuiyan, M.M.H.; Hossain, M.R.; Bashar, H.; Rahaman, M.S.; Manir, M.S.; Ullah, S.M.; Uddin, S.S.; Mollah, M.Z.I.; Khan, R.A. Improvement of efficiency of Dye Sensitized Solar Cells by optimizing the combination ratio of Natural Red and Yellow dyes. Optik 2019, 179, 252–258. [Google Scholar] [CrossRef]
- Guzel, E.; Arslan, B.S.; Durmaz, V.; Cesur, M.; Tutar, O.F.; Sari, T.; Isleyen, M.; Nebioglu, M.; Sisman, I. Photovoltaic performance and photostability of anthocyanins, isoquinoline alkaloids and betalains as natural sensitizers for DSSCs. Sol. Energy 2018, 173, 34–41. [Google Scholar] [CrossRef]
- Boyo, A.O.; Boyo, H.O.; Shitta, M.B.O.; Abudusalam, T.I.; Fajana, O.O.; Awodibo, M.A. Fabrication of dye-sensitized solar cells (dssc) device using lawsonia inermis leaf. Int. J. Pure Appl. Phys. 2013, 2, 59–68. [Google Scholar]
- Prabavathy, N.; Shalini, S.; Balasundaraprabhu, R.; Velauthapillai, D.; Prasanna, S.; Walke, P.; Muthukumarasamy, N. Effect of solvents in the extraction and stability of anthocyanin from the petals of Caesalpinia pulcherrima for natural dye sensitized solar cell applications. J. Mater. Sci. Mater. Electron. 2017, 28, 9882–9892. [Google Scholar] [CrossRef]
- Rajkumar, S.; Kumar, M.N.; Suguna, K.; Muthulakshmi, S.; Kumar, R.A. Enhanced performance of dye-sensitized solar cells using natural cocktail dye as sensitizer. Optik 2019, 178, 224–230. [Google Scholar] [CrossRef]
- Zhu, W.H.; Wu, Y.Z.; Wang, S.T.; Li, W.Q.; Li, X.; Chen, J.; Wang, Z.S.; Tian, H. Organic D-A-π-A Solar Cell Sensitizers with Improved Stability and Spectral Response. Adv. Funct. Mater. 2011, 21, 756–763. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1697–A1705. [Google Scholar] [CrossRef]
- Sanusi, K.; Fatomi, N.O.; Borisade, A.O.; Yilmaz, Y.; Ceylan, Ü.; Fashina, A. An approximate procedure for profiling dye molecules with potentials as sensitizers in solar cell application: A DFT/TD-DFT approach. Chem. Phys. Lett. 2019, 723, 111–117. [Google Scholar] [CrossRef]
- Sadki, H.; Bourass, M.; Bennani, M.N.; Bouachrine, M. New organic materials based on D-π-A structure for application in dye-sensitized solar cells. Res. Chem. Intermed. 2018, 44, 6071–6085. [Google Scholar] [CrossRef]
- El-Meligy, A.B.; Koga, N.; Iuchi, S.; Yoshida, K.; Hirao, K.; Mangood, A.H.; El-Nahas, A.M. DFT/TD-DFT calculations of the electronic and optical properties of bis-N,N-dimethylaniline-based dyes for use in dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2018, 367, 332–346. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Zai, J.; Ma, D.; Hu, Z.; He, Q.; Wu, M.; Chen, D.; Chen, Z.; Qian, X. Improving the catalytic performance of Ni3S4-PtCo heteronanorods via Mott-Schottky effect toward the reduction of iodine couples in dye-sensitized solar cells. Electrochim. Acta. 2017, 241, 89–97. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.B.; Li, Y.Z.; Song, P.; Ma, F.C.; Sun, M.T. Photoactive layer based on T-shaped benzimidazole dyes used for solar cell: From photoelectric properties to molecular design. Sci. Rep. 2017, 7, 45688. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, Version 3.1; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Galappaththi, K.; Ekanayake, P.; Petra, M.I. A rational design of high efficient and low-cost dye sensitizer with exceptional absorptions: Computational study of cyanidin based organic sensitizer. Sol. Energy 2018, 161, 83–89. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Wang, L.X.; Liu, Y.; Tuo, X.L.; Li, S.N.; Wang, M.G. Effect of H+ and NH4+ on the N-NO2 bond dissociation energy of HMX. Acta Phys.Chim. Sin. 2007, 23, 1560–1564. [Google Scholar]
- Shi, X.L.; Yang, Y.H.; Wang, L.H.; Li, Y.Z. Introducing Asymmetry Induced by Benzene Substitution in a Rigid Fused pi Spacer of D-pi-A-Type Solar Cells: A Computational Investigation. J. Phys. Chem. C 2019, 123, 4007–4021. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Ramasamy, E.; Lee, J. Ferrocene-derivatized ordered mesoporous carbon as high performance counter electrodes for dye-sensitized solar cells. Carbon 2010, 48, 3715–3720. [Google Scholar] [CrossRef]
- De Leeuw, D.M.; Simenon, M.M.J.; Brown, A.R.; Einerhand, R.E.F. Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met. 1997, 87, 53–59. [Google Scholar] [CrossRef]
- Cahen, D.; Hodes, G.; Gratzel, M.; Guillemoles, J.F.; Riess, I. Nature of Photovoltaic Action in Dye-Sensitized Solar Cells. J. Phys. Chem. B 2000, 104, 2053–2059. [Google Scholar] [CrossRef]
- Asbury, J.B.; Wang, Y.Q.; Hao, E.; Ghosh, H.N.; Lian, T. Evidences of hot excited state electron injection from sensitizer molecules to TiO2 nanocrystalline thin films. Res. Chem. Intermed. 2001, 27, 393–406. [Google Scholar] [CrossRef]
- Zhan, C.G.; Nichols, J.A.; Dixon, D.A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity Index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; Van Speybroeck, V.; Waroquier, M.; Geerlings, P.; De Proft, F. Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. [Google Scholar] [CrossRef]
- Ruhle, S.; Greenshtein, M.; Chen, S.G.; Merson, A.; Pizem, H.; Sukenik, C.S.; Cahen, D.; Zaban, A. Molecular adjustment of the electronic properties of nanoporous electrodes in dye-sensitized solar cells. J. Phys. Chem. B 2005, 109, 18907–18913. [Google Scholar] [CrossRef] [PubMed]
- Fahim, Z.M.E.; Bouzzine, S.M.; Youssef, A.A.; Bouachrine, M.; Hamidi, M. Ground state geometries, UV/vis absorption spectra and charge transfer properties of triphenylamine-thiophenes based dyes for DSSCs: A TD-DFT benchmark study. Comput. Theor. Chem. 2018, 1125, 39–48. [Google Scholar] [CrossRef]
- German, E.; Faccio, R.; Mombrú, A.W. Comparison of standard DFT and Hubbard-DFT methods in structural and electronic properties of TiO2 polymorphs and H-titanate ultrathin sheets for DSSC application. Appl. Surf. Sci. 2018, 428, 118–123. [Google Scholar] [CrossRef]
- Li, Y.Z.; Li, Y.C.; Song, P.; Ma, F.C.; Liang, J.P.; Sun, M.T. Screening and design of high-performance indoline-based dyes for DSSCs. RSC Adv. 2017, 7, 20520–20536. [Google Scholar] [CrossRef] [Green Version]
- Sheela, N.R.; Muthu, S.; Sampathkrishnan, S. Molecular orbital studies (hardness, chemical potential and electrophilicity), vibrational investigation and theoretical NBO analysis of 4-4-(1H-1,2,4-triazol-1-yl methylene) dibenzonitrile based on abinitio and DFT methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.F.; Li, Y.; Han, J.H.; Cao, B.F.; Yin, H.; Shi, Y. Enhanced photoelectrical properties of alizarin-based natural dye via structure modulation. Sol. Energy 2019, 185, 315–323. [Google Scholar] [CrossRef]
- Le Bahers, T.; Adamo, C.; Ciofini, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Peng, X.P.; Song, F.S.; Lu, E.; Wang, Y.N.; Zhou, W.; Fan, J.L.; Gao, Y.L. Heptamethine Cyanine Dyes with a Large Stokes Shift and Strong Fluorescence: A Paradigm for Excited-State Intramolecular Charge Transfer. J. Am. Chem. Soc. 2005, 127, 4170–4171. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Chen, Y.; Gou, G.Z.; Mu, W.H.; Lv, X.J.; Du, M.L.; Fu, W.F. Large Stokes Shift Induced by Intramolcular Charge Transfer in N,O-Chelated Naphthyridine–BF2 Complexes. Org. Lett. 2012, 14, 5226–5229. [Google Scholar] [CrossRef]
- Estrella, L.L.; Balanay, M.P.; Kim, D.H. The Effect of Donor Group Rigidification on the Electronic and Optical Properties of Arylamine-Based Metal-Free Dyes for Dye-Sensitized Solar Cells: A Computational Study. J. Phys. Chem. A 2016, 120, 5917–5927. [Google Scholar] [CrossRef]
- Mu, X.J.; Wang, J.G.; Sun, M.T. Visualization of Photoinduced Charge Transfer and Electron–Hole Coherence in Two-Photon Absorption. J. Phys. Chem. C 2019, 123, 14132–14143. [Google Scholar] [CrossRef]
- Geng, H.; Peng, Q.; Wang, L.J.; Li, H.J.; Liao, Y.; Ma, Z.Y.; Shuai, Z.G. Toward Quantitative Prediction of Charge Mobility in Organic Semiconductors: Tunneling Enabled Hopping Model. Adv. Mater. 2012, 24, 3568–3572. [Google Scholar] [CrossRef] [PubMed]
- Le Bahers, T.; Pauporte, T.; Scalmani, G.; Adamo, C.; Ciofini, I. A TD-DFT investigation of ground and excited state properties in indoline dyes used for dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2009, 11, 11276–11284. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, S.; Bahadur, L. Enhancement of power conversion efficiency of dye-sensitized solar cells by co-sensitization of Phloxine B and Bromophenol blue dyes on ZnO photoanode. J. Lumin 2015, 161, 426–430. [Google Scholar] [CrossRef]
- Li, Y.Z.; Xu, B.B.; Song, P.; Ma, F.C.; Sun, M.T. D-A-π-A System: Light Harvesting, Charge Transfer, and Molecular Designing. J. Phys. Chem. C 2017, 121, 12546–12561. [Google Scholar] [CrossRef]
- Jeon, I.Y.; Kim, H.M.; Kweon, D.H.; Jung, S.M.; Seo, J.M.; Shin, S.H.; Choi, I.T.; Eom, Y.K.; Kang, S.H.; Kim, H.K.; et al. Metalloid tellurium-doped graphene nanoplatelets as ultimately stable electrocatalysts for cobalt reduction reaction in dye-sensitized solar cells. Nano Energy 2016, 30, 867–876. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.B.; Sun, S.L.; Geng, Y.; Wu, Y.; Su, Z.M. Density functional theory characterization and design of high-performance diarylamine-fluorene dyes with different π spacers for dye-sensitized solar cells. J. Mater. Chem. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Ren, P.H.; Sun, C.F.; Shi, Y.; Song, P.; Yang, Y.H.; Li, Y.Z. Global performance evaluation of solar cells using two models: From charge-transfer and recombination mechanisms to photoelectric properties. J. Mater. Chem. C 2019, 7, 1934–1947. [Google Scholar] [CrossRef]
- Wang, Z.S.; Cui, Y.; Hara, K.; Dan-Oh, Y.; Kasada, C.; Shinpo, A. A high-light-N harvesting-efficiency coumarin dye for stable dye-sensitized solar cells. Adv. Mater. 2007, 19, 1138–1141. [Google Scholar] [CrossRef]
- Bao, C.; Li, F.; Wang, J.; Sun, P.; Huang, N.; Sun, Y.; Fang, L.; Wang, L.; Sun, X. One-Pot Solvothermal in Situ Growth of 1D Single-Crystalline NiSe on Ni Foil as Efficient and Stable Transparent Conductive Oxide Free Counter Electrodes for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2016, 8, 32788–32796. [Google Scholar] [CrossRef]
- Urbani, M.; Sari, F.A.; Graetzel, M.; Nazeeruddin, M.K.; Torres, T.; Ince, M. Effect of Peripheral Substitution on the Performance of Subphthalocyanines in DSSCs. Chem. Asian J. 2016, 11, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Sugihara, H.; Arakawa, H. Molecular design of ruthenium(II) polypyridyl photosensitizers for efficient nanocrystalline TiO2 solar cells. J. Photochem. Photobiol. A Chem. 2003, 158, 131–138. [Google Scholar] [CrossRef]
- Ardo, S.; Meyer, G.J. Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces. Chem. Soc. Rev. 2009, 38, 115–164. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.J.; Fu, Y.; Tian, H. Improvement of dye-sensitized solar cells: What we know and what we need to know. Energy Environ. Sci. 2010, 3, 1170–1181. [Google Scholar] [CrossRef]
- Preat, J.; Jacquemin, D.; Perpète, E.A. Towards new efficient dye-sensitised solar cells. Energy Environ. Sci. 2010, 3, 891–904. [Google Scholar] [CrossRef]
- Kleinman, D.A. Nonlinear Dielectric Polarization in Optical Media. Phys. Rev. 1962, 126, 1977–1979. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dyes | S-Q (eV) | S-R (eV) |
|---|---|---|
| −5.834 | −5.907 | |
| −1.862 | −1.715 | |
| 3.972 | 4.192 |
| Dyes | S-Q | S-R |
|---|---|---|
| IP (eV) | 5.482 | 5.264 |
| EA (eV) | 2.164 | 2.058 |
| W (eV) | 0.576 | 0.571 |
| W− (eV) | 6.524 | 6.211 |
| W+ (eV) | 2.701 | 2.550 |
| X(eV) | 3.823 | 3.661 |
| H(eV) | 1.659 | 1.603 |
| S(eV−1) | 1.652 | 1.642 |
| Ф(eV) | 12.123 | 10.742 |
| Dyes | States | E (eV) | λ (nm) | CI | F |
|---|---|---|---|---|---|
| S-R | S1 | 3.6800 | 336.92 | H→L/0.682 | 0.3600 |
| S2 | 3.7822 | 327.81 | H-1→L/0.655 | 0.0104 | |
| S-Q | S1 | 3.4876 | 355.50 | H→L/0.675 | 0.4010 |
| S2 | 3.7928 | 326.89 | H-1→L/0.678 | 0.1096 |
| Dyes | ||
|---|---|---|
| S-Q | 0.6861 | 0.5321 |
| S-R | 0.8812 | 0.5932 |
| Dyes | (mAcm−2) | (V) | ff | η (%) | |
|---|---|---|---|---|---|
| S-Q | 5.480 | 0.582 | 0.674 | 0.344 | 2.151 |
| S-R | 1.826 | 0.547 | 0.714 | 0.114 | 0.713 |
| Dyes | LHE | |||
|---|---|---|---|---|
| S-Q | 0.603 | 5.834 | 2.347 | −1.653 |
| S-R | 0.563 | 5.907 | 2.227 | −1.773 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Lu, Q.; Su, R.; Li, Y.; Zhao, M. Light Harvesting and Optical-Electronic Properties of Two Quercitin and Rutin Natural Dyes. Appl. Sci. 2019, 9, 2567. https://doi.org/10.3390/app9122567
Zhao D, Lu Q, Su R, Li Y, Zhao M. Light Harvesting and Optical-Electronic Properties of Two Quercitin and Rutin Natural Dyes. Applied Sciences. 2019; 9(12):2567. https://doi.org/10.3390/app9122567
Chicago/Turabian StyleZhao, Dongpeng, Qiuchen Lu, Runzhou Su, Yuanzuo Li, and Meiyu Zhao. 2019. "Light Harvesting and Optical-Electronic Properties of Two Quercitin and Rutin Natural Dyes" Applied Sciences 9, no. 12: 2567. https://doi.org/10.3390/app9122567
APA StyleZhao, D., Lu, Q., Su, R., Li, Y., & Zhao, M. (2019). Light Harvesting and Optical-Electronic Properties of Two Quercitin and Rutin Natural Dyes. Applied Sciences, 9(12), 2567. https://doi.org/10.3390/app9122567