Insights into the Variations of Hao-Dependent Nitrifying and Nir-Dependent Denitrifying Microbial Communities in Ammonium-Graduated Lake Environments


Abstract
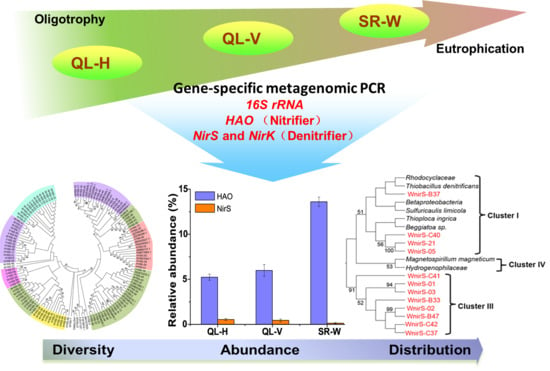
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Chemical Analyses
2.2. Primer Design and PCR Amplification
2.3. Quantitative PCR Analysis of hao, nirS, nirK, and 16S rDNA Genes
2.4. Phylogeny Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Result
3.1. Environmental Characteristics
3.2. Distributional Differences in Microbial Communities in Nitrogen-Related Environments
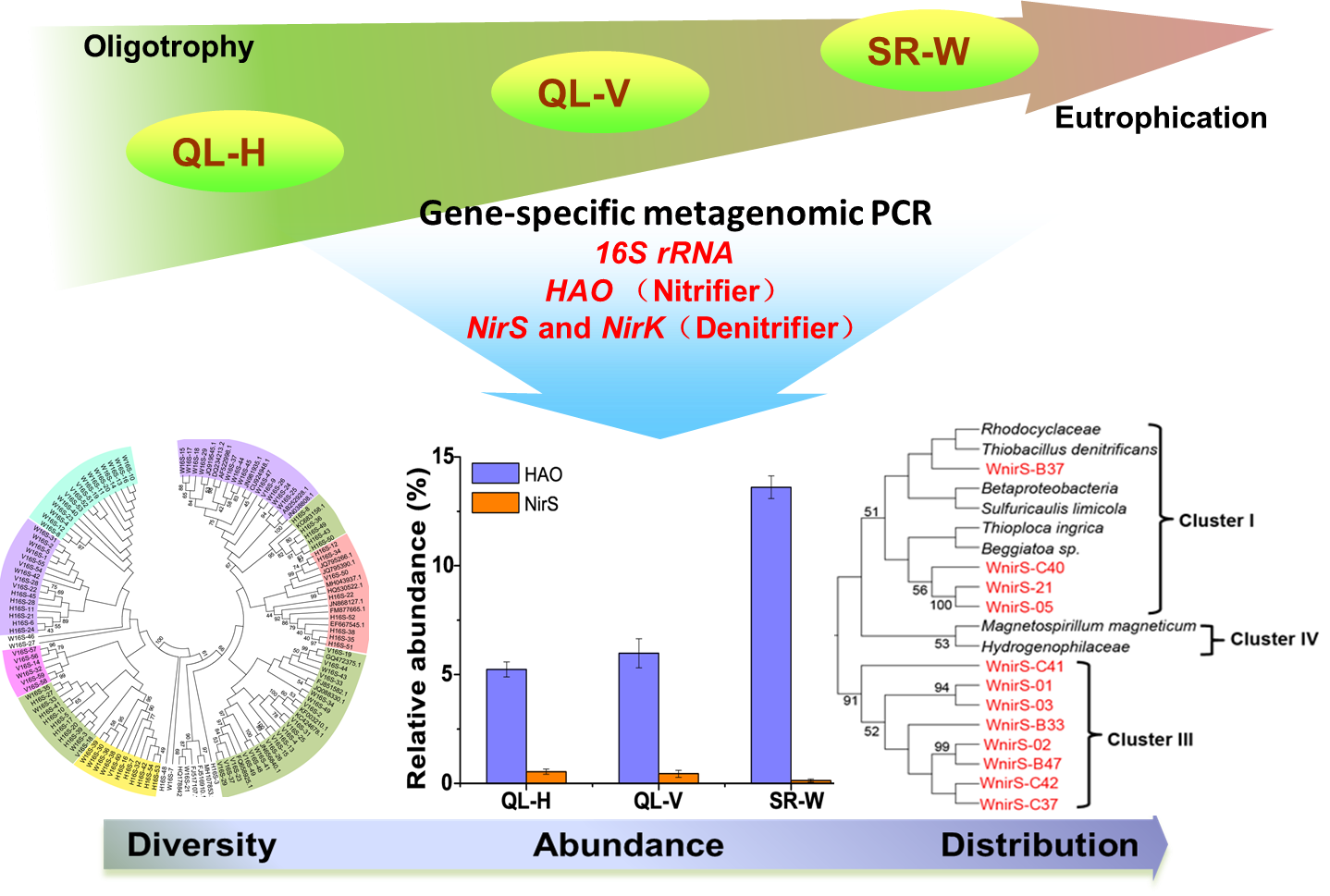
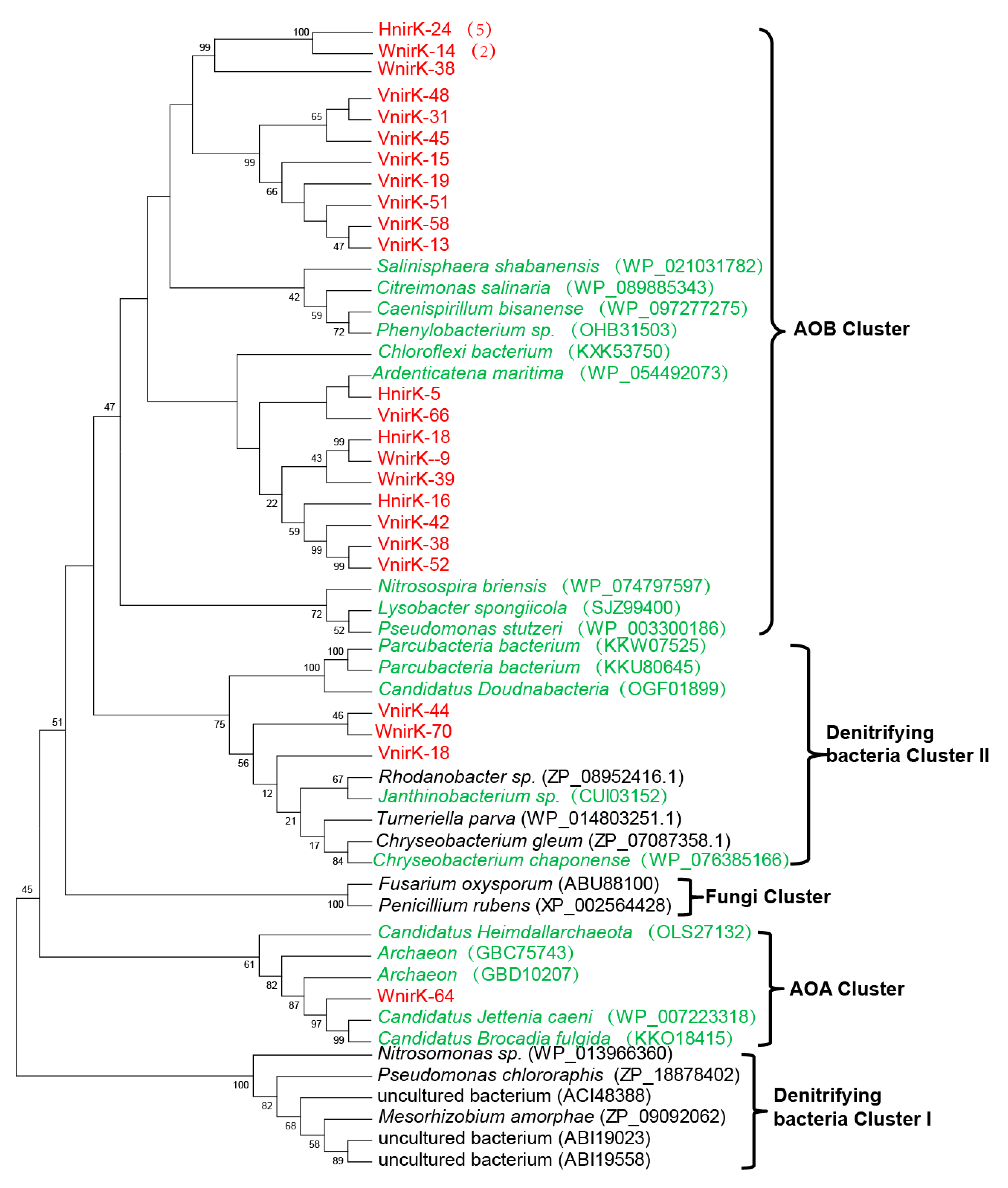
3.3. Phylogenetic Diversity of Hao-Dependent Nitrifying Bacteria in Sediments
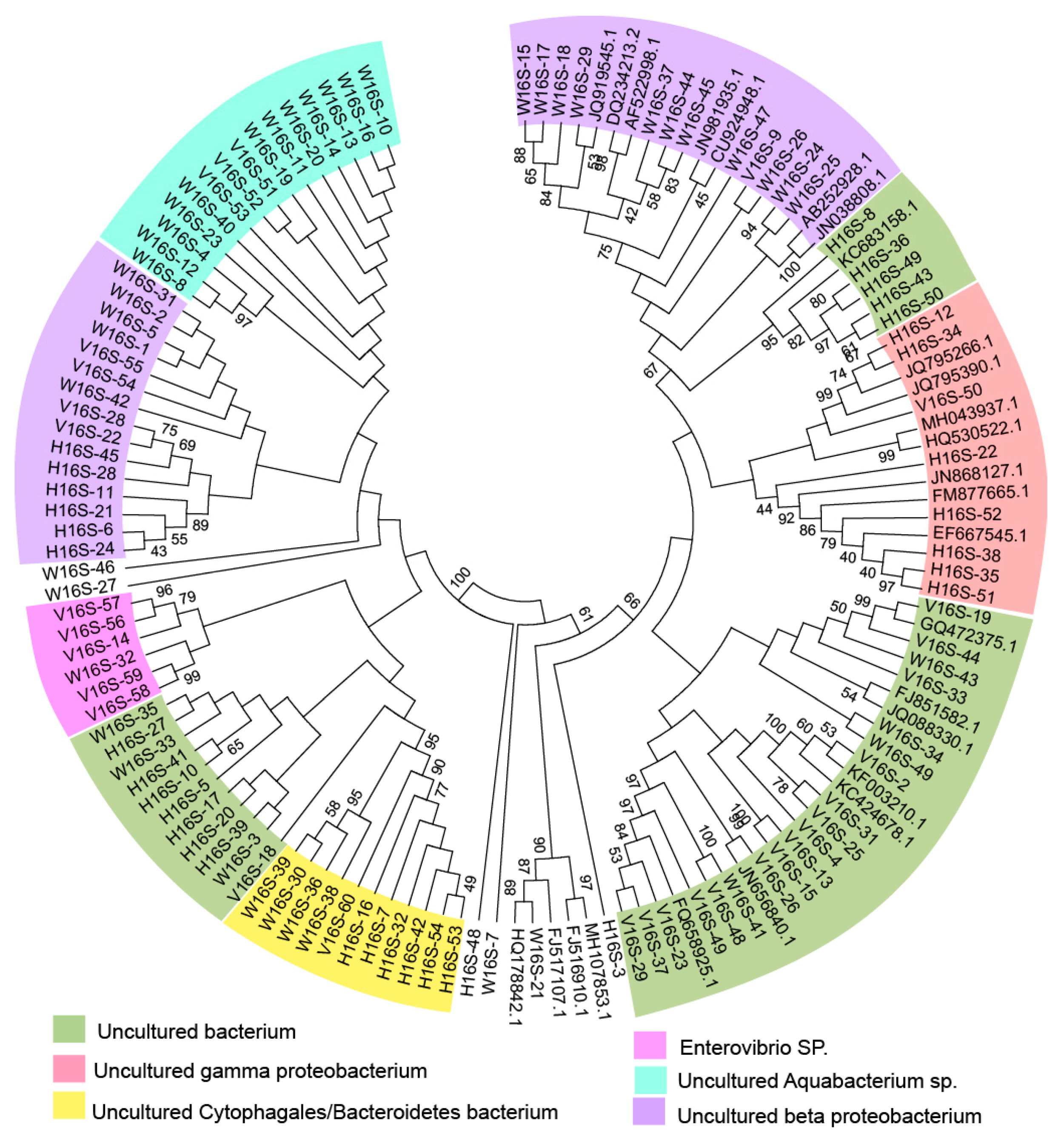
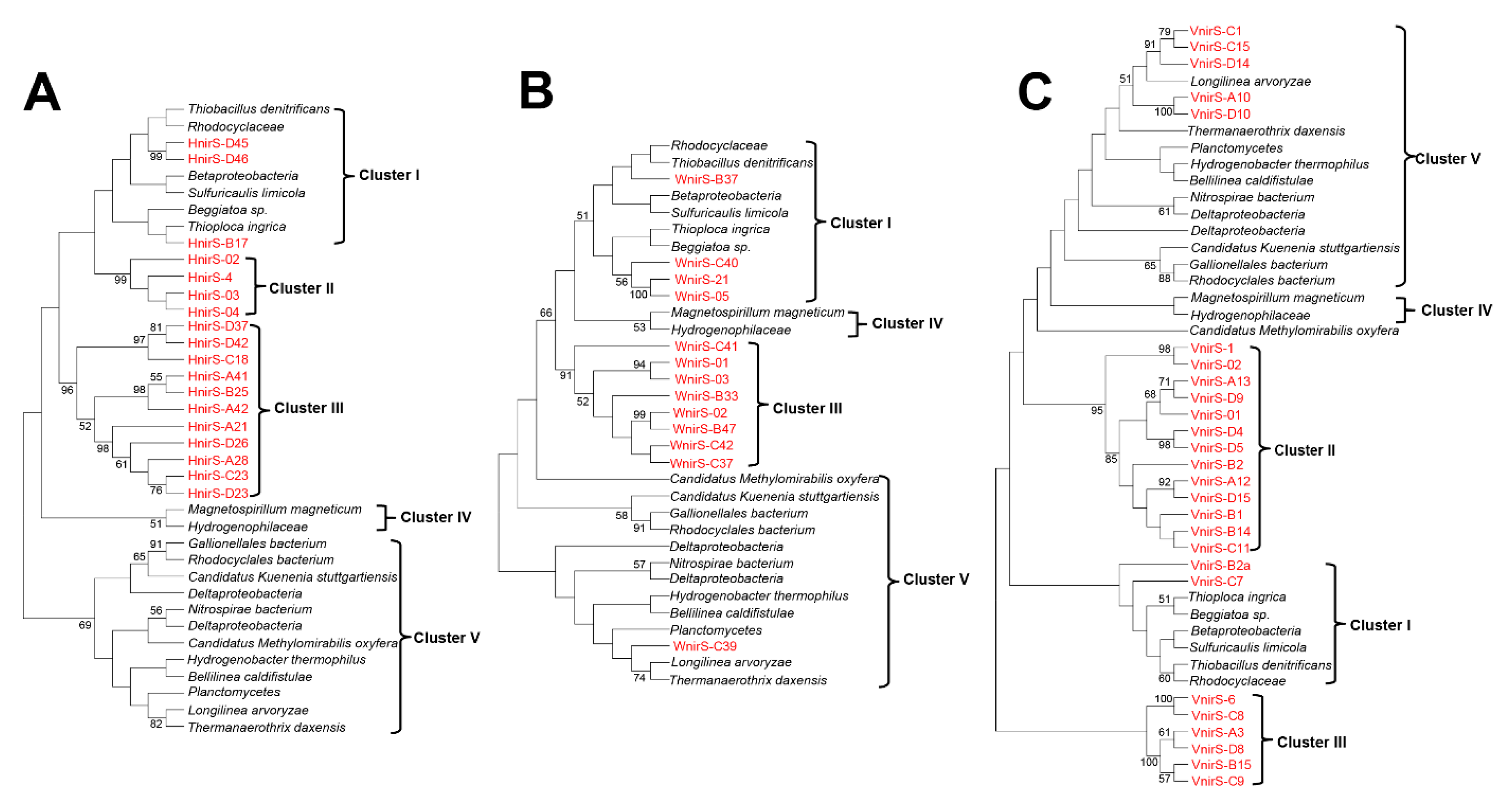
3.4. Diversity of NirS-Dependent Denitrifying Communities
3.5. Diversity of NirK-Dependent Denitrifying Communities
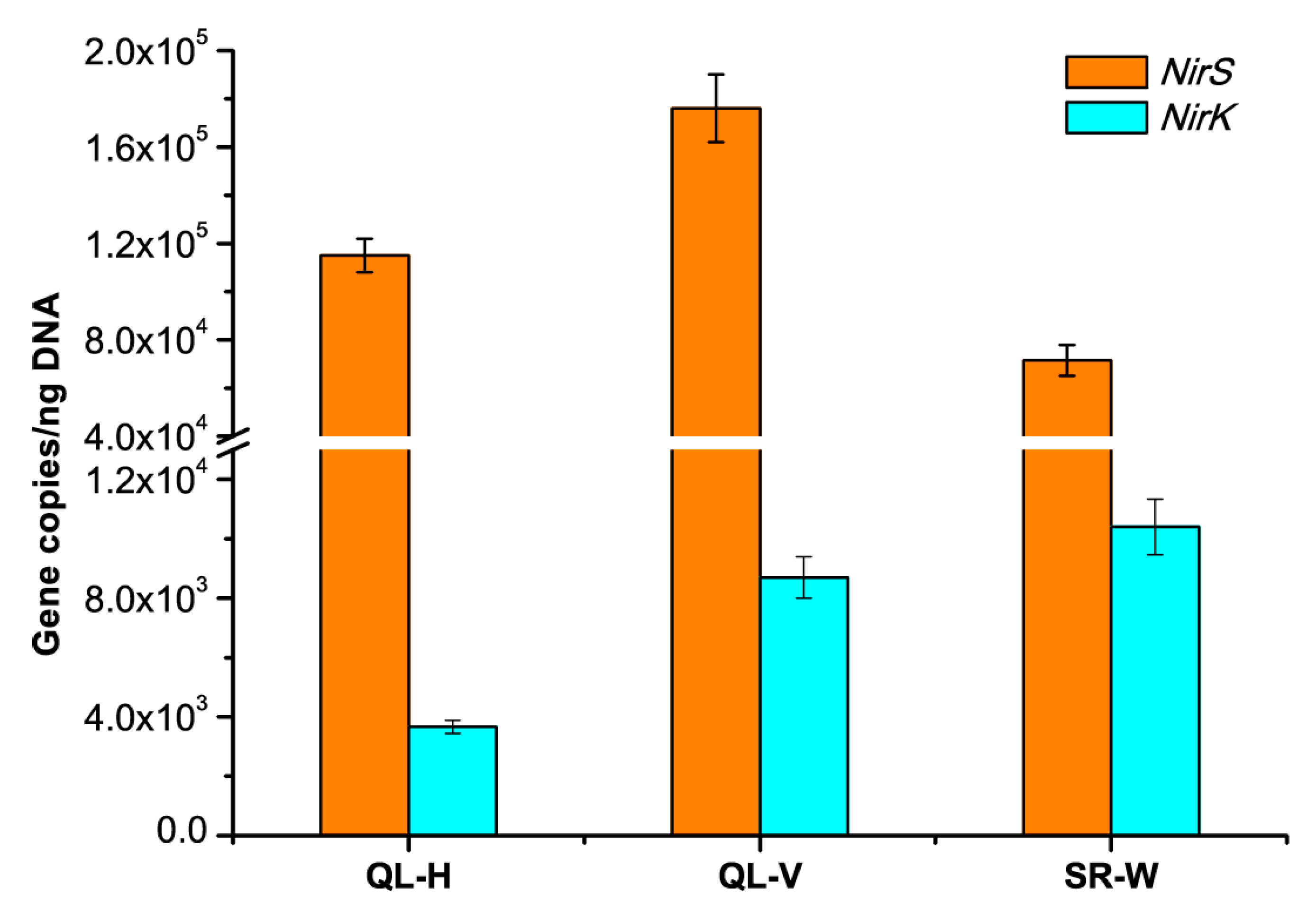
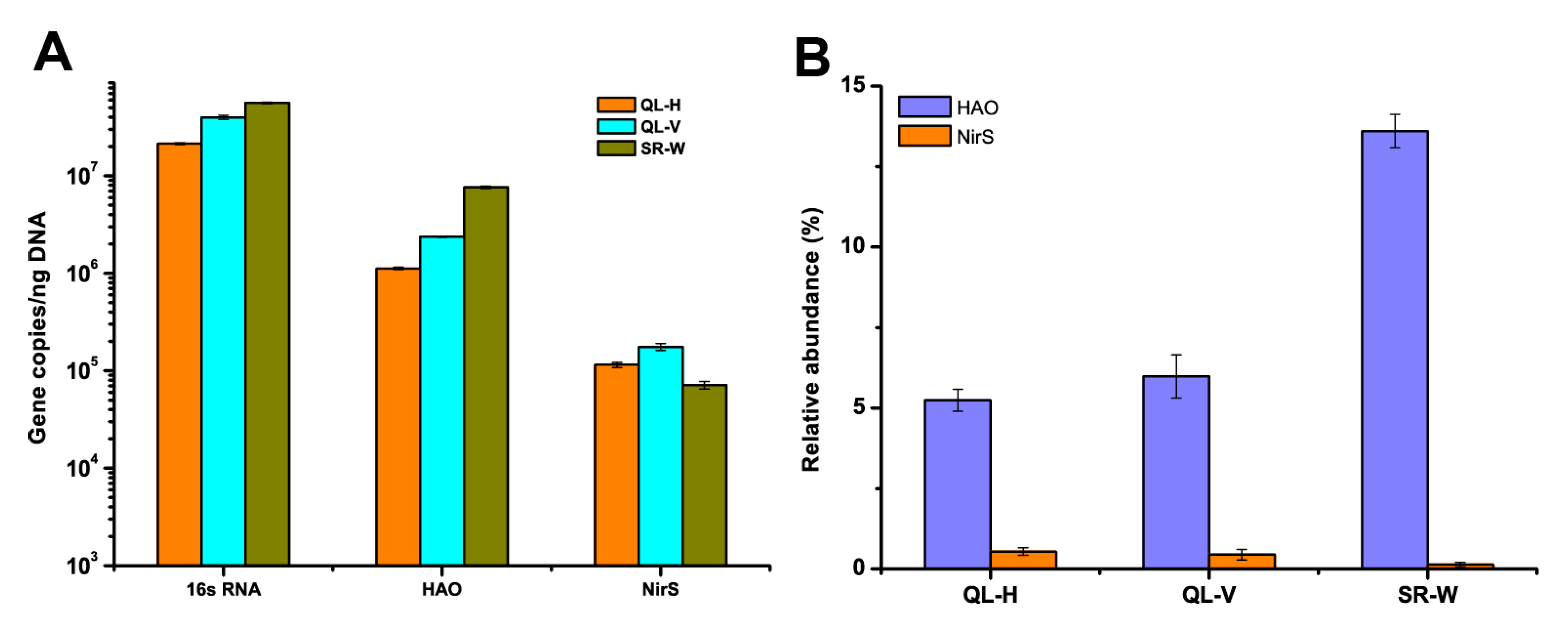
3.6. Abundances of Nitrifying and Denitrifying Bacterial Communities
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Philippot, L.; Hallin, S.; Schloter, M. Ecology of denitrifying prokaryotes in agricultural soil. Adv. Agron. 2007, 96, 249–305. [Google Scholar]
- Arrigo, K.R. Marine microorganisms and global nutrient cycles. Nature 2005, 437, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wan, S. Independent effects of warming and nitrogen addition on plant phenology in the Inner Mongolian steppe. Ann. Bot. Lond. 2013, 111, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, J.; Sun, G.E.; Chu, H.; Noormets, A.; Ouyang, Z.; Ranjeet, J.; Wan, S.; Guan, W. Long-term variability and environmental control of the carbon cycle in an oak-dominated temperate forest. For. Ecol. Manag. 2014, 313, 319–328. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hao, L.; Zhang, Q.; Dai, S. Effect of nitrite on growth and microcystins production of Microcystis aeruginosa PCC7806. J. Appl. Phycol. 2011, 23, 665–671. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, X.; Jiao, N.; Hsiao, S.S.Y.; Kao, S.J. Diversity and distribution of amoA-type nitrifying and nirS-type denitrifying microbial communities in the Yangtze River estuary. Biogeosciences 2014, 12, 2131–2145. [Google Scholar] [CrossRef]
- Oakley, B.B.; Francis, C.A.; Roberts, K.J.; Fuchsman, C.A.; Srinivasan, S.; Staley, J.T. Analysis of nitrite reductase (nirK and nirS) genes and cultivation reveal depauperate community of denitrifying bacteria in the Black Sea suboxic zone. Environ. Microbiol. 2007, 9, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hou, L.; Zheng, Y.; Liu, M.; Yin, G.; Li, X.; Lin, X.; Yu, C.; Wang, R.; Jiang, X. Nirs-Encoding denitrifier community composition, distribution, and abundance along the coastal wetlands of China. Appl. Microbiol. Biotechnol. 2016, 100, 8573–8582. [Google Scholar] [CrossRef]
- Zhang, T.; Ye, L.; Tong, A.H.; Shao, M.F.; Lok, S. Ammonia-oxidizing archaea and ammonia-oxidizing bacteria in six full-scale wastewater treatment bioreactors. Appl. Microbiol. Biotechnol. 2011, 91, 1215–1225. [Google Scholar] [CrossRef] [Green Version]
- Figuerola, E.L.M.; Erijman, L. Diversity of nitrifying bacteria in a full-scale petroleum refinery wastewater treatment plant experiencing unstable nitrification. J. Hazard. Mater. 2010, 181, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.X.; Lu, X.; Liu, B.; Li, Y.; Long, C.; Li, A. Abundance and diversity of bacterial nitrifiers and denitrifiers and their functional genes in tannery wastewater treatment plants revealed by high-throughput sequencing. PLoS ONE 2014, 9, e113603. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Bao, Q.; Jirimutu; Qing, M.; Siriguleng; Chen, X.; Sun, T.; Li, M.; Zhang, J.; Yu, J. Isolation and identification of lactic acid bacteria from Tarag in Eastern Inner Mongolia of China by 16S rRNA sequences and DGGE analysis. Microbiol. Res. 2012, 167, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.S.; Agterveld, K.V.; Zwart, G. Distribution of typical freshwater bacterial groups is associated with ph, temperature, and lake water retention time. Appl. Environ. Microb. 2005, 71, 8201–8206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Fu, S.; Mathew, R.P.; Lawrence, K.S.; Feng, Y. Soil microbial community structure and activity in a 100-year-old fertilization and crop rotation experiment. J. Plant Ecol. 2015, 60, 741–747. [Google Scholar] [CrossRef]
- Iwai, S.; Chai, B.; Sul, W.J.; Cole, J.R.; Hashsham, S.A.; Tiedje, J.M. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 2010, 4, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Pei, X.; Du, P.; Yin, X.; Xiong, X.; Wu, H.; Zhou, X.; Wang, Q. Overexpression and characterization of a new organic solvent-tolerant esterase derived from soil metagenomic DNA. Bioresour. Technol. 2012, 116, 234–240. [Google Scholar] [CrossRef]
- Montgomery, H.A.C.; Thom, N.S.; Cockburn, A. Determination of dissolved oxygen by the winkler method and the solubility of oxygen in pure water and sea water. J. Chem. Technol. Biot. 2010, 14, 280–296. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Regan, J.M.; Harrington, G.W.; Noguera, D.R. Ammonia- and nitrite-oxidizing bacterial communities in a pilot-scale chloraminated drinking water distribution system. Appl. Environ. Microb. 2002, 68, 73–81. [Google Scholar] [CrossRef]
- Ball, P.N.; Mackenzie, M.D.; Deluca, T.H.; Holben, W.E. Wildfire and charcoal enhance nitrification and ammonium-oxidizing bacterial abundance in dry montane forest soils. J. Environ. Qual. 2010, 39, 1243–1253. [Google Scholar] [CrossRef]
- Kartal, B.; Maalcke, W.J.; Almeida, N.M.D.; Cirpus, I.; Gloerich, J.; Geerts, W.; Camp, H.J.M.O.D.; Harhangi, H.R.; Janssenmegens, E.M.; Francoijs, K.J. Molecular mechanism of anaerobic ammonium oxidation. Nature 2011, 479, 127–130. [Google Scholar] [CrossRef]
- Dang, H.; Wang, C.; Li, J.; Li, T.; Tian, F.; Jin, W.; Ding, Y.; Zhang, Z. Diversity and distribution of sediment nirS-encoding bacterial assemblages in response to environmental gradients in the eutrophied Jiaozhou Bay, China. Microb. Ecol. 2010, 58, 161–169. [Google Scholar] [CrossRef]
- Long, A.; Song, B.; Fridey, K.; Silva, A. Detection and diversity of copper containing nitrite reductase genes (nirK) in prokaryotic and fungal communities of agricultural soils. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef]
- Shen, L.D.; Liu, S.; Zhu, Q.; Li, X.Y.; Cai, C.; Cheng, D.Q.; Lou, L.P.; Xu, X.Y.; Zheng, P.; Hu, B.L. Distribution and diversity of nitrite-dependent anaerobic methane-oxidising bacteria in the sediments of the Qiantang River. Microb. Ecol. 2014, 67, 341–349. [Google Scholar] [CrossRef]
- Gao, D.; Wang, X.; Fu, S.; Zhao, J. Legume plants enhance the resistance of soil to ecosystem disturbance. Front. Plant Sci. 2017, 8, 1295. [Google Scholar] [CrossRef]
- Zhang, X.; Li, A.; Szewzyk, U.; Fang, M. Improvement of biological nitrogen removal with nitrate-dependent Fe(II) oxidation bacterium Aquabacterium parvum B6 in an up-flow bioreactor for wastewater treatment. Bioresour. Technol. 2016, 219, 624–631. [Google Scholar] [CrossRef]
- Eiler, A.; Langenheder, S.; Bertilsson, S.; Tranvik, L.J. Heterotrophic bacterial growth efficiency and community structure at different natural organic carbon concentrations. Appl. Environ. Microb. 2003, 69, 3701–3709. [Google Scholar] [CrossRef]
- Fernándezgómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrósalió, C. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ford, T.; Li, X.; Gu, J.D. Cytochrome cd1-containing nitrite reductase encoding gene nirS as a new functional biomarker for detection of anaerobic ammonium oxidizing (Anammox) bacteria. Environ. Sci. Technol. 2011, 45, 3547–3553. [Google Scholar] [CrossRef]
- Quan, Z.X.; Rhee, S.K.; Zuo, J.E.; Yang, Y.; Bae, J.W.; Park, J.R.; Lee, S.T.; Park, Y.H. Diversity of ammonium-oxidizing bacteria in a granular sludge anaerobic ammonium-oxidizing (anammox) reactor. Environ. Microbiol. 2008, 10, 3130–3139. [Google Scholar] [CrossRef]
- Gieseke, A.; Bjerrum, L.; Wagner, M.; Amann, R. Structure and activity of multiple nitrifying bacterial populations co-existing in a biofilm. Environ. Microbiol. 2003, 5, 355–369. [Google Scholar] [CrossRef]
- Gieseke, A.; Purkhold, U.; Wagner, M.; Amann, R.; Schramm, A. Community structure and activity dynamics of nitrifying bacteria in a phosphate-removing biofilm. Appl. Environ. Microb. 2001, 67, 1351–1362. [Google Scholar] [CrossRef]
- Allen, J.G.; Beutel, M.W.; Call, D.R.; Fischer, A.M. Effects of oxygenation on ammonia oxidation potential and microbial diversity in sediment from surface-flow wetland mesocosms. Bioresour. Technol. 2010, 101, 1389–1392. [Google Scholar] [CrossRef]
- Gabriele, S.; Barbara, B.; Petra, D.; Gabriele, R.; Koops, H.P. The ammonia-oxidizing nitrifying population of the River Elbe estuary. FEMS Microbiol. Ecol. 2010, 17, 177–186. [Google Scholar]
- Bollmann, A.; Laanbroek, H.J. Continuous culture enrichments of ammonia-oxidizing bacteria at low ammonium concentrations. FEMS Microbiol. Ecol. 2001, 37, 211–221. [Google Scholar] [CrossRef]
- Gorshkova, N.M.; Ivanova, E.P.; Sergeev, A.F.; Zhukova, N.V.; Alexeeva, Y.; Wright, J.P.; Nicolau, D.V.; Mikhailov, V.V.; Christen, R. Marinobacter excellens sp. nov., isolated from sediments of the Sea of Japan. Int. J. Syst. Evol. Micr. 2003, 53, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Torrentó, C.; Urmeneta, J.; Otero, N.; Soler, A.; Viñas, M.; Cama, J. Enhanced denitrification in groundwater and sediments from a nitrate-contaminated aquifer after addition of pyrite. Chem. Geol. 2011, 287, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zheng, Y.; Wu, S.; Yang, Z.H.; Zhao, F. Bacterial community structure of autotrophic denitrification biocathode by 454 pyrosequencing of the 16s rrna gene. Microb. Ecol. 2015, 69, 492–499. [Google Scholar] [CrossRef]
- Hernsdorf, A.W.; Amano, Y.; Miyakawa, K.; Ise, K.; Suzuki, Y.; Anantharaman, K.; Probst, A.; Burstein, D.; Thomas, B.C.; Banfield, J.F. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017, 11, 1915–1929. [Google Scholar] [CrossRef]
- Kim, Y.C.; Gao, C.; Zheng, Y.; He, X.H.; Yang, W.; Chen, L.; Wan, S.Q.; Guo, L.D. Arbuscular mycorrhizal fungal community response to warming and nitrogen addition in a semiarid steppe ecosystem. Mycorrhiza 2015, 25, 267. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hao, L. Intracellular nitrite accumulation: The cause of growth inhibition of Microcystis aeruginosa exposure to high nitrite level. Phycol. Res. 2015, 63, 197–201. [Google Scholar] [CrossRef]
- Mosier, A.C.; Francis, C.A. Denitrifier abundance and activity across the San Francisco Bay estuary. Environ. Microbiol. Rep. 2010, 2, 667–676. [Google Scholar] [CrossRef]
- Nogales, B.; Timmis, K.; Nedwell, D.; Osborn, A. Detection and diversity of expressed denitrification genes in estuarine sediments after reverse transcription-PCR amplification from mRNA. Appl. Environ. Microb. 2002, 68, 5017–5025. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TN | NH4+-N | NO3−-N | NO2−-N | COD | pH | DO | |
|---|---|---|---|---|---|---|---|
| QL-H | 0.21 | 0.08 | 0.06 | ND a | 13 | 6.83 | 4.91 |
| QL-V | 16.82 | 14.36 | 1.15 | 0.14 | 75 | 6.97 | 5.71 |
| SR-W | 121.32 | 93.41 | 5.67 | 0.87 | 331 | 7.71 | 3.35 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Chen, Y.; Qu, J.; Jin, P.; Zheng, Z.; Cui, Z. Insights into the Variations of Hao-Dependent Nitrifying and Nir-Dependent Denitrifying Microbial Communities in Ammonium-Graduated Lake Environments. Appl. Sci. 2019, 9, 3229. https://doi.org/10.3390/app9163229
Zhao R, Chen Y, Qu J, Jin P, Zheng Z, Cui Z. Insights into the Variations of Hao-Dependent Nitrifying and Nir-Dependent Denitrifying Microbial Communities in Ammonium-Graduated Lake Environments. Applied Sciences. 2019; 9(16):3229. https://doi.org/10.3390/app9163229
Chicago/Turabian StyleZhao, Ruojin, Yinyan Chen, Jin Qu, Peng Jin, Zhanwang Zheng, and Zhiwen Cui. 2019. "Insights into the Variations of Hao-Dependent Nitrifying and Nir-Dependent Denitrifying Microbial Communities in Ammonium-Graduated Lake Environments" Applied Sciences 9, no. 16: 3229. https://doi.org/10.3390/app9163229
APA StyleZhao, R., Chen, Y., Qu, J., Jin, P., Zheng, Z., & Cui, Z. (2019). Insights into the Variations of Hao-Dependent Nitrifying and Nir-Dependent Denitrifying Microbial Communities in Ammonium-Graduated Lake Environments. Applied Sciences, 9(16), 3229. https://doi.org/10.3390/app9163229