Equivalence of RABBITT and Streaking Delays in Attosecond-Time-Resolved Photoemission Spectroscopy at Solid Surfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
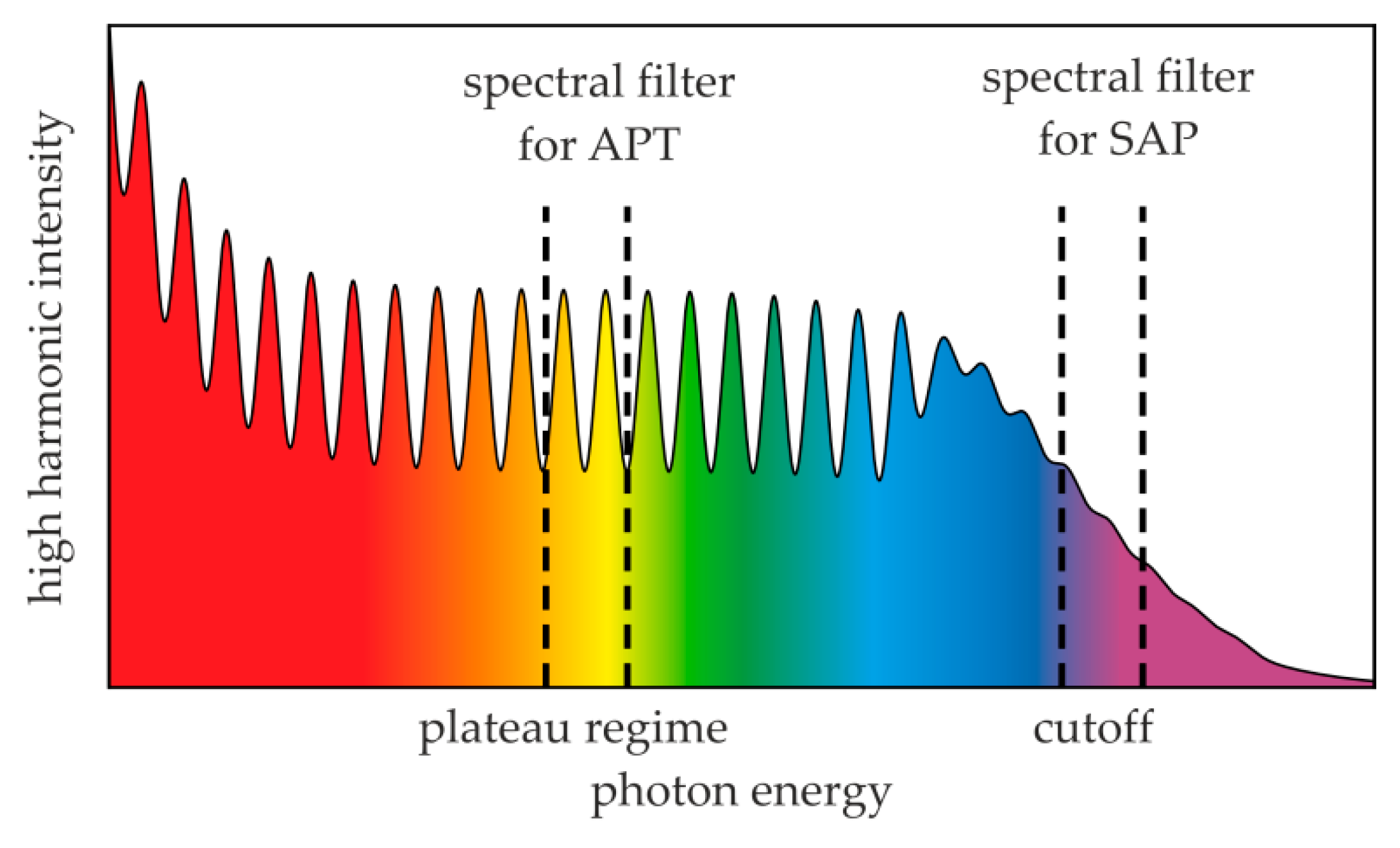
:1. Introduction
2. Methods
3. Results
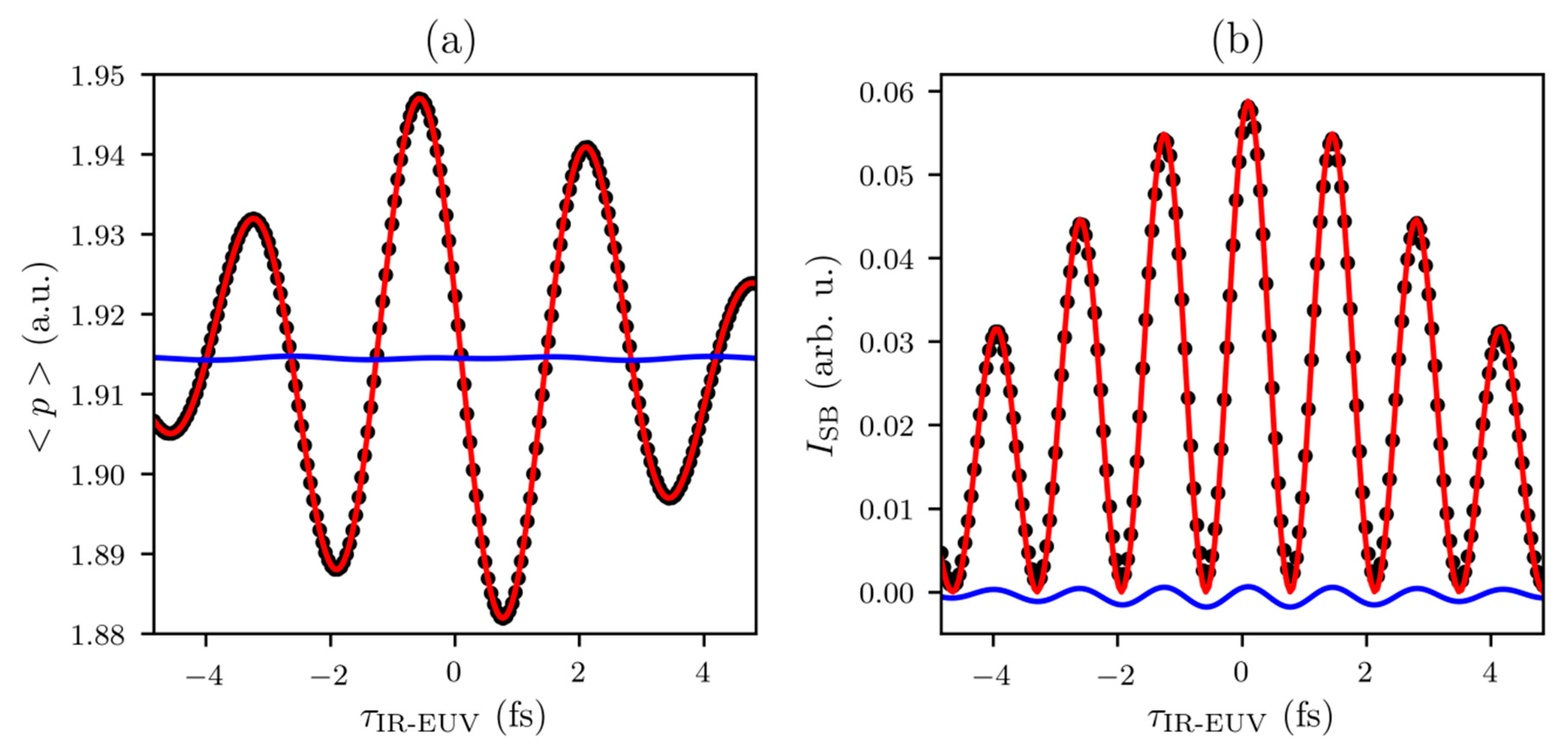
3.1. From RABBITT Regime to Streaking Regime
3.2. Comparison of Streaking and RABBITT Delays with Classical Photoemission Times
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Extraction of Delays

References
- Drescher, M.; Hentschel, M.; Kienberger, R.; Tempea, G.; Spielmann, C.; Reider, G.A.; Corkum, P.B.; Krausz, F. X-ray Pulses Approaching the Attosecond Frontier. Science 2001, 291, 1923–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, P.M.; Toma, E.S.; Breger, P.; Mullot, G.; Augé, F.; Balcou, P.; Muller, H.G.; Agostini, P. Observation of a Train of Attosecond Pulses from High Harmonic Generation. Science 2001, 292, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, M.; Kienberger, R.; Spielmann, C.; Reider, G.A.; Milosevic, N.; Brabec, T.; Corkum, P.; Heinzmann, U.; Drescher, M.; Krausz, F. Attosecond metrology. Nature 2001, 414, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Gallmann, L.; Cirelli, C.; Keller, U. Attosecond Science: Recent Highlights and Future Trends. Annu. Rev. Phys. Chem. 2012, 63, 447–469. [Google Scholar] [PubMed]
- Schultze, M.; Fiess, M.; Karpowicz, N.; Gagnon, J.; Korbman, M.; Hofstetter, M.; Neppl, S.; Cavalieri, A.L.; Komninos, Y.; Mercouris, T.; et al. Delay in Photoemission. Science 2010, 328, 1658–1662. [Google Scholar] [CrossRef] [PubMed]
- Klünder, K.; Dahlström, J.M.; Gisselbrecht, M.; Fordell, T.; Swoboda, M.; Guénot, D.; Johnsson, P.; Caillat, J.; Mauritsson, J.; Maquet, A.; et al. Probing Single-Photon Ionization on the Attosecond Time Scale. Phys. Rev. Lett. 2011, 106, 143002. [Google Scholar] [CrossRef] [PubMed]
- Dahlström, J.M.; Guénot, D.; Klünder, K.; Gisselbrecht, M.; Mauritsson, J.; L’Huillier, A.; Maquet, A.; Taïeb, R. Theory of attosecond delays in laser-assisted photoionization. Chem. Phys. 2013, 414, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, M.; Decleva, P.; Calegari, F.; Palacios, A.; Martín, F. Attosecond Electron Dynamics in Molecules. Chem. Rev. 2017, 117, 10760–10825. [Google Scholar] [CrossRef]
- Neppl, S.; Ernstorfer, R.; Cavalieri, A.L.; Lemell, C.; Wachter, G.; Magerl, E.; Bothschafter, E.M.; Jobst, M.; Hofstetter, M.; Kleineberg, U.; et al. Direct observation of electron propagation and dielectric screening on the atomic length scale. Nature 2015, 517, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Siek, F.; Neb, S.; Bartz, P.; Hensen, M.; Strüber, C.; Fiechter, S.; Torrent-Sucarrat, M.; Silkin, V.M.; Krasovskii, E.E.; Kabachnik, N.M.; et al. Angular momentum–induced delays in solid-state photoemission enhanced by intra-atomic interactions. Science 2017, 357, 1274–1277. [Google Scholar] [CrossRef]
- Ossiander, M.; Riemensberger, J.; Neppl, S.; Mittermair, M.; Schäffer, M.; Duensing, A.; Wagner, M.S.; Heider, R.; Wurzer, M.; Gerl, M.; et al. Absolute timing of the photoelectric effect. Nature 2018, 561, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, A.L.; Müller, N.; Uphues, T.; Yakovlev, V.S.; Baltuška, A.; Horvath, B.; Schmidt, B.; Blümel, L.; Holzwarth, R.; Hendel, S.; et al. Attosecond spectroscopy in condensed matter. Nature 2007, 449, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, L.; Liu, Q.; Zherebtsov, S.; Trabattoni, A.; Rupp, P.; Castrovilli, M.C.; Galli, M.; Süßmann, F.; Wintersperger, K.; Stierle, J.; et al. Attosecond chronoscopy of electron scattering in dielectric nanoparticles. Nat. Phys. 2017, 13, 766–770. [Google Scholar] [CrossRef]
- Pazourek, R.; Nagele, S.; Burgdörfer, J. Attosecond chronoscopy of photoemission. Rev. Mod. Phys. 2015, 87, 765–802. [Google Scholar] [CrossRef] [Green Version]
- Maquet, A.; Caillat, J.; Taïeb, R. Attosecond delays in photoionization: time and quantum mechanics. J. Phys. B At. Mol. Opt. Phys. 2014, 47, 204004. [Google Scholar] [CrossRef]
- Kazansky, A.K.; Echenique, P.M. One-Electron Model for the Electronic Response of Metal Surfaces to Subfemtosecond Photoexcitation. Phys. Rev. Lett. 2009, 102, 177401. [Google Scholar] [CrossRef] [PubMed]
- Lemell, C.; Solleder, B.; Tőkési, K.; Burgdörfer, J. Simulation of attosecond streaking of electrons emitted from a tungsten surface. Phys. Rev. A 2009, 79, 062901. [Google Scholar] [CrossRef]
- Krasovskii, E.E.; Friedrich, C.; Schattke, W.; Echenique, P.M. Rapid propagation of a Bloch wave packet excited by a femtosecond ultraviolet pulse. Phys. Rev. B 2016, 94, 195434. [Google Scholar] [CrossRef] [Green Version]
- Borisov, A.G.; Sánchez-Portal, D.; Kazansky, A.K.; Echenique, P.M. Resonant and nonresonant processes in attosecond streaking from metals. Phys. Rev. B 2013, 87, 121110. [Google Scholar] [CrossRef]
- Krasovskii, E.E. Attosecond spectroscopy of solids: Streaking phase shift due to lattice scattering. Phys. Rev. B 2011, 84, 195106. [Google Scholar] [CrossRef]
- Liao, Q.; Thumm, U. Attosecond Time-Resolved Photoelectron Dispersion and Photoemission Time Delays. Phys. Rev. Lett. 2014, 112, 023602. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, A.; Neb, S.; Enns, W.; Heinzmann, U.; Kazansky, A.K.; Pfeiffer, W. Photoemission time versus streaking delay in attosecond time-resolved solid state photoemission. Submitted to EPJ Web Conf. 2018. [Google Scholar]
- Tao, Z.; Chen, C.; Szilvasi, T.; Keller, M.; Mavrikakis, M.; Kapteyn, H.; Murnane, M. Direct time-domain observation of attosecond final-state lifetimes in photoemission from solids. Science 2016, 353, 62–67. [Google Scholar] [CrossRef]
- Smirnova, O.; Mouritzen, A.S.; Patchkovskii, S.; Ivanov, M.Y. Coulomb–laser coupling in laser-assisted photoionization and molecular tomography. J. Phys. B At. Mol. Opt. Phys. 2007, 40, F197–F206. [Google Scholar] [CrossRef] [Green Version]
- Nagele, S.; Pazourek, R.; Feist, J.; Doblhoff-Dier, K.; Lemell, C.; Tőkési, K.; Burgdörfer, J. Time-resolved photoemission by attosecond streaking: extraction of time information. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 081001. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; L’Huillier, A.; Ferray, M.; Lompré, L.A.; Mainfray, G. Multiple-harmonic generation in rare gases at high laser intensity. Phys. Rev. A 1989, 39, 5751–5761. [Google Scholar] [CrossRef]
- McPherson, A.; Gibson, G.; Jara, H.; Johann, U.; Luk, T.S.; McIntyre, I.A.; Boyer, K.; Rhodes, C.K. Studies of multiphoton production of vacuum-ultraviolet radiation in the rare gases. J. Opt. Soc. Am. B 1987, 4, 595. [Google Scholar] [CrossRef]
- Muller, H.G. Reconstruction of attosecond harmonic beating by interference of two-photon transitions. Appl. Phys. B 2002, 74, s17–s21. [Google Scholar] [CrossRef]
- Itatani, J.; Quéré, F.; Yudin, G.L.; Ivanov, M.Y.; Krausz, F.; Corkum, P.B. Attosecond Streak Camera. Phys. Rev. Lett. 2002, 88, 173903. [Google Scholar] [CrossRef] [Green Version]
- Miaja-Avila, L.; Lei, C.; Aeschlimann, M.; Gland, J.L.; Murnane, M.M.; Kapteyn, H.C.; Saathoff, G. Laser-Assisted Photoelectric Effect from Surfaces. Phys. Rev. Lett. 2006, 97, 113604. [Google Scholar] [CrossRef]
- Cattaneo, L.; Vos, J.; Lucchini, M.; Gallmann, L.; Cirelli, C.; Keller, U. Comparison of attosecond streaking and RABBITT. Opt. Express 2016, 24, 29060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasmi, L.; Lucchini, M.; Castiglioni, L.; Kliuiev, P.; Osterwalder, J.; Hengsberger, M.; Gallmann, L.; Krüger, P.; Keller, U. Effective mass effect in attosecond electron transport. Optica 2017, 4, 1492. [Google Scholar] [CrossRef]
- Kazansky, A.K.; Kabachnik, N.M. Theoretical description of atomic photoionization by an attosecond XUV pulse in a strong laser field: effects of rescattering and orbital polarization. J. Phys. B At. Mol. Opt. Phys. 2007, 40, 2163–2177. [Google Scholar] [CrossRef]
- Fleck, J.A.; Morris, J.R.; Feit, M.D. Time-Dependent Propagation of High Energy Laser Beams through the Atmosphere. Appl. Phys. B 1976, 10, 129–160. [Google Scholar] [CrossRef]
- Feit, M.D.; Fleck, J.A. Solution of the Schrödinger equation by a spectral method II: Vibrational energy levels of triatomic molecules. J. Chem. Phys. 1983, 78, 301–308. [Google Scholar] [CrossRef]
- Lewenstein, M.; Balcou, P.; Ivanov, M.Y.; L’Huillier, A.; Corkum, P.B. Theory of high-harmonic generation by low-frequency laser fields. Phys. Rev. A 1994, 49, 2117–2132. [Google Scholar] [CrossRef] [PubMed]
- Corkum, P.B. Plasma perspective on strong field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [Green Version]
- Seah, M.P.; Dench, W.A. Quantitative Electron Spectroscopy of Surfaces: A Standard Data Base for Electron Inelastic Mean Free Paths in Solids. Surf. Interface Anal. 1979, 1, 2–11. [Google Scholar] [CrossRef]
- Guggenmos, A.; Akil, A.; Ossiander, M.; Schäffer, M.; Azzeer, A.M.; Boehm, G.; Amann, M.-C.; Kienberger, R.; Schultze, M.; Kleineberg, U. Attosecond photoelectron streaking with enhanced energy resolution for small-bandgap materials. Opt. Lett. 2016, 41, 3714. [Google Scholar] [CrossRef]
- Chen, C.; Tao, Z.; Carr, A.; Matyba, P.; Szilvási, T.; Emmerich, S.; Piecuch, M.; Keller, M.; Zusin, D.; Eich, S.; et al. Distinguishing attosecond electron–electron scattering and screening in transition metals. Proc. Natl. Acad. Sci. USA 2017, 114, E5300–E5307. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Galán, Á.; Martín, F.; Argenti, L. Two-photon finite-pulse model for resonant transitions in attosecond experiments. Phys. Rev. A 2016, 93, 023429. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebauer, A.; Neb, S.; Enns, W.; Stadtmüller, B.; Aeschlimann, M.; Pfeiffer, W. Equivalence of RABBITT and Streaking Delays in Attosecond-Time-Resolved Photoemission Spectroscopy at Solid Surfaces. Appl. Sci. 2019, 9, 592. https://doi.org/10.3390/app9030592
Gebauer A, Neb S, Enns W, Stadtmüller B, Aeschlimann M, Pfeiffer W. Equivalence of RABBITT and Streaking Delays in Attosecond-Time-Resolved Photoemission Spectroscopy at Solid Surfaces. Applied Sciences. 2019; 9(3):592. https://doi.org/10.3390/app9030592
Chicago/Turabian StyleGebauer, Andreas, Sergej Neb, Walter Enns, Benjamin Stadtmüller, Martin Aeschlimann, and Walter Pfeiffer. 2019. "Equivalence of RABBITT and Streaking Delays in Attosecond-Time-Resolved Photoemission Spectroscopy at Solid Surfaces" Applied Sciences 9, no. 3: 592. https://doi.org/10.3390/app9030592
APA StyleGebauer, A., Neb, S., Enns, W., Stadtmüller, B., Aeschlimann, M., & Pfeiffer, W. (2019). Equivalence of RABBITT and Streaking Delays in Attosecond-Time-Resolved Photoemission Spectroscopy at Solid Surfaces. Applied Sciences, 9(3), 592. https://doi.org/10.3390/app9030592