Analytical Theory of Attosecond Transient Absorption Spectroscopy of Perturbatively Dressed Systems


Abstract
:1. Introduction
2. Formal Analysis
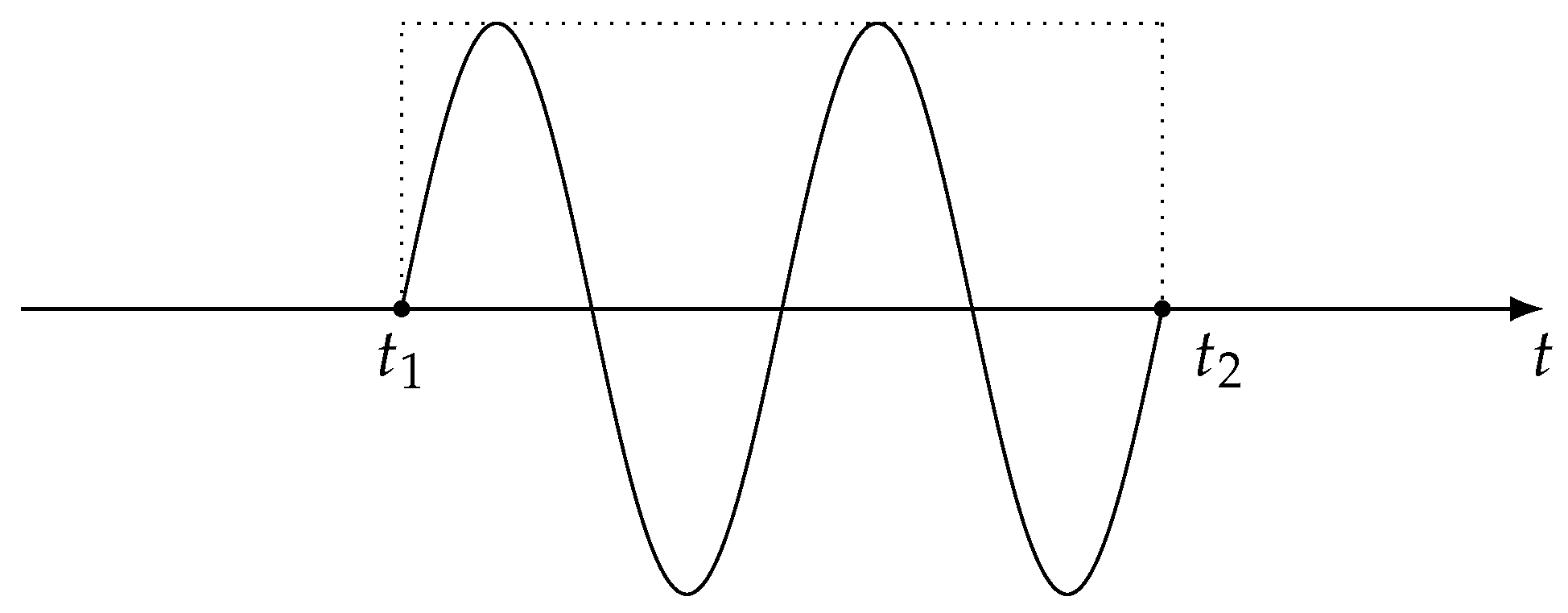
2.1. XUV One-Photon Absorption Cross-Section
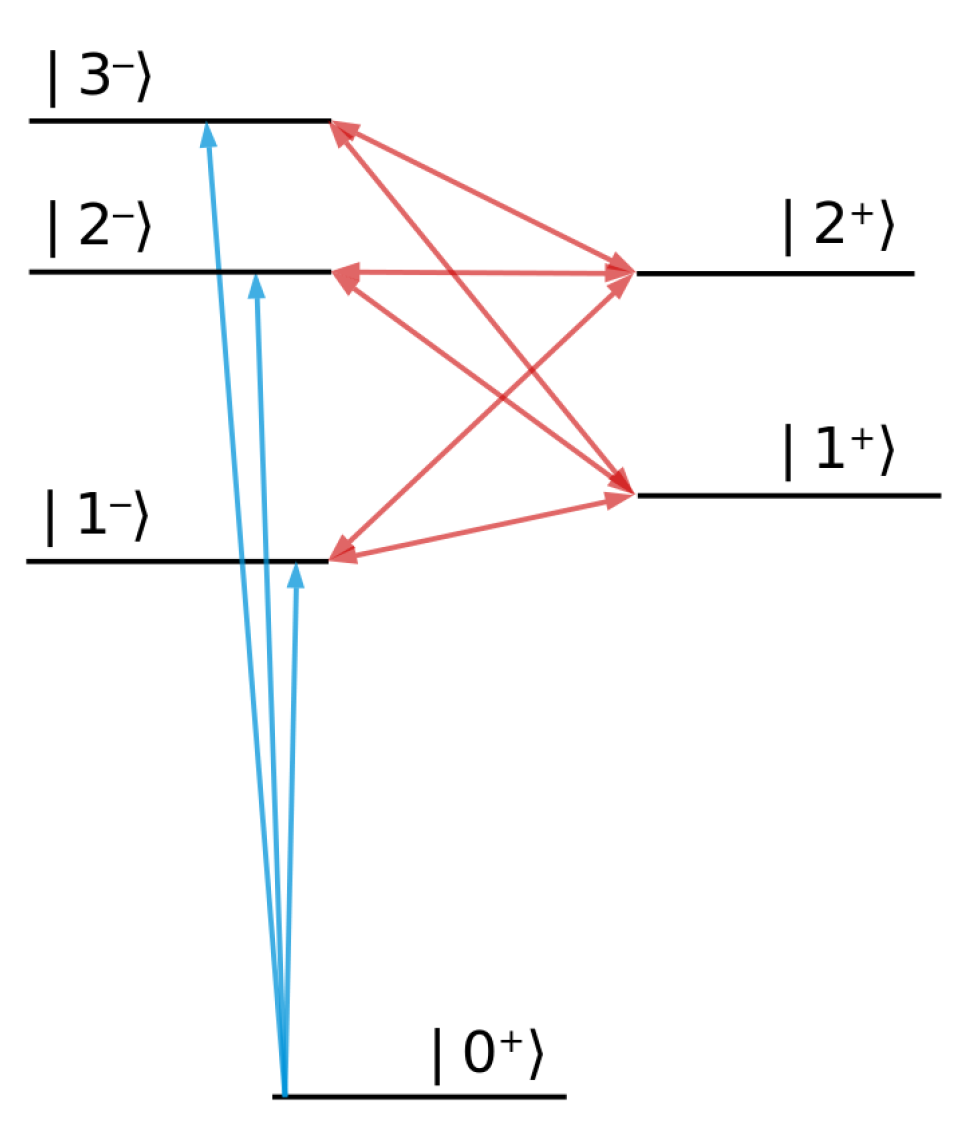
2.2. Perturbative Treatment of the Dressing Pulse
2.3. Analytical Structure of the Cross-Section
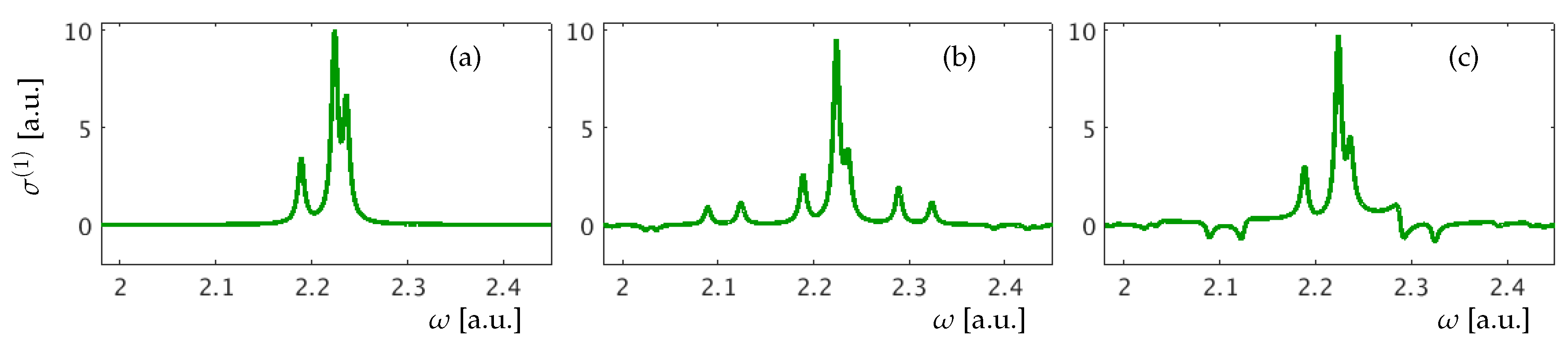
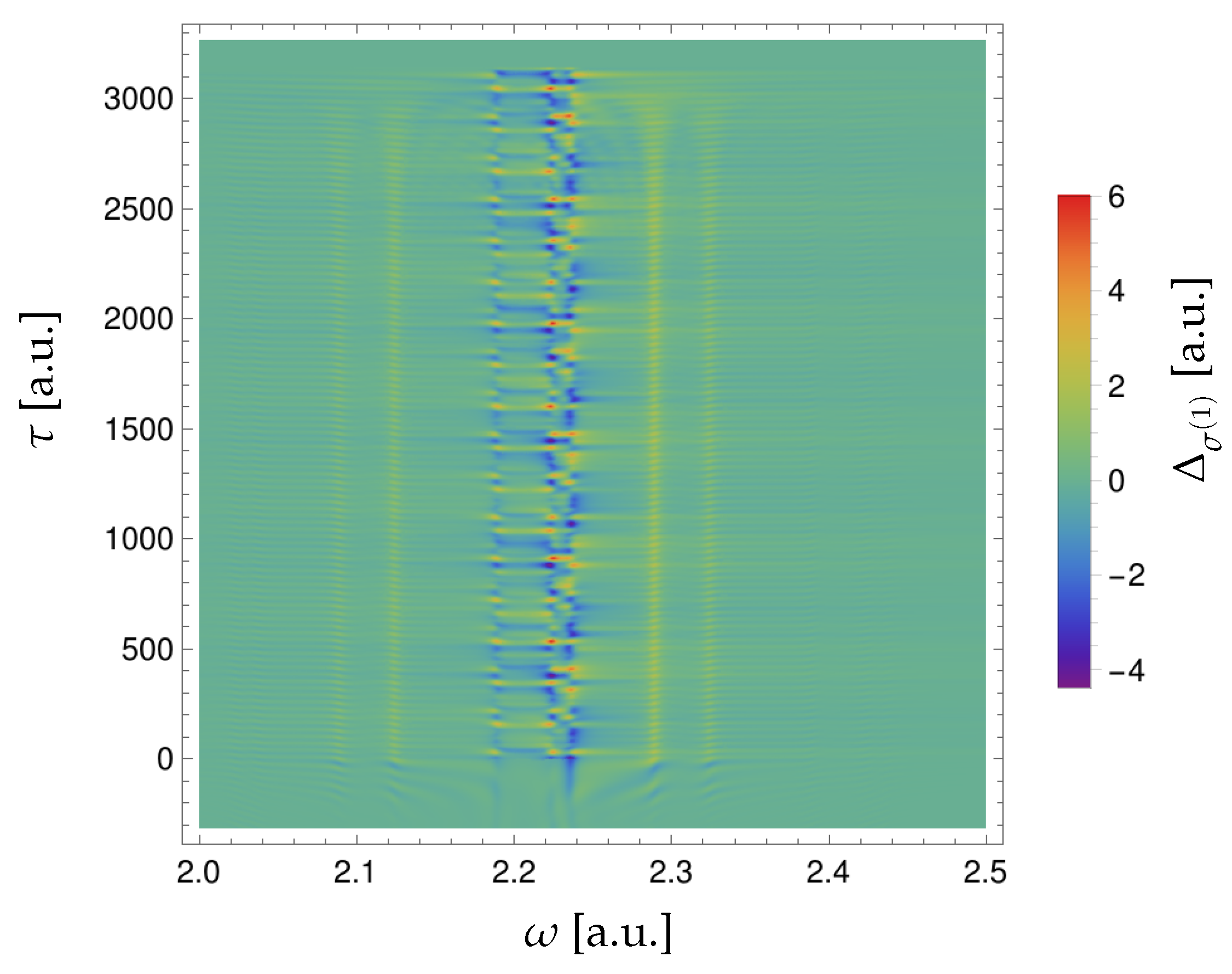
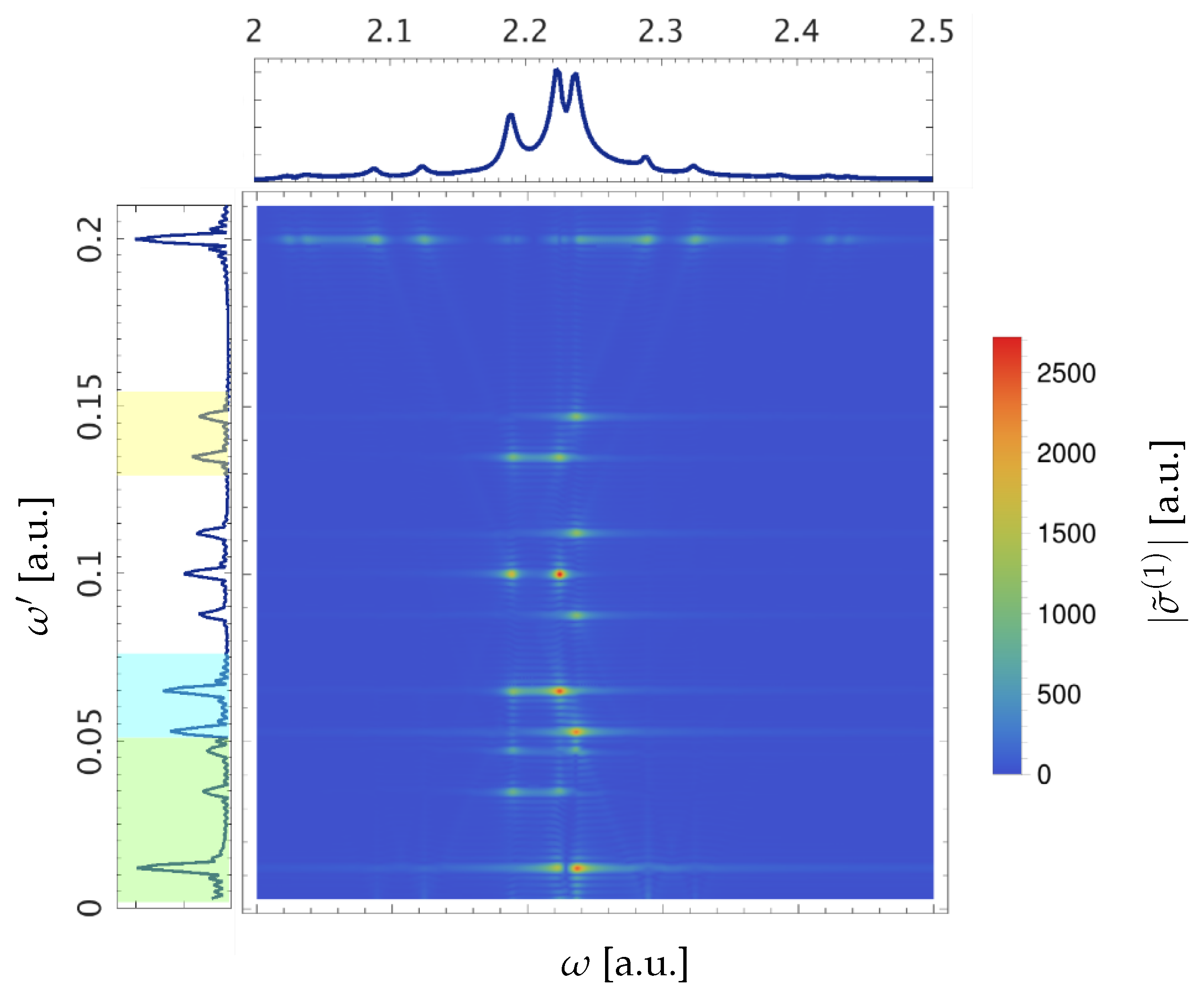
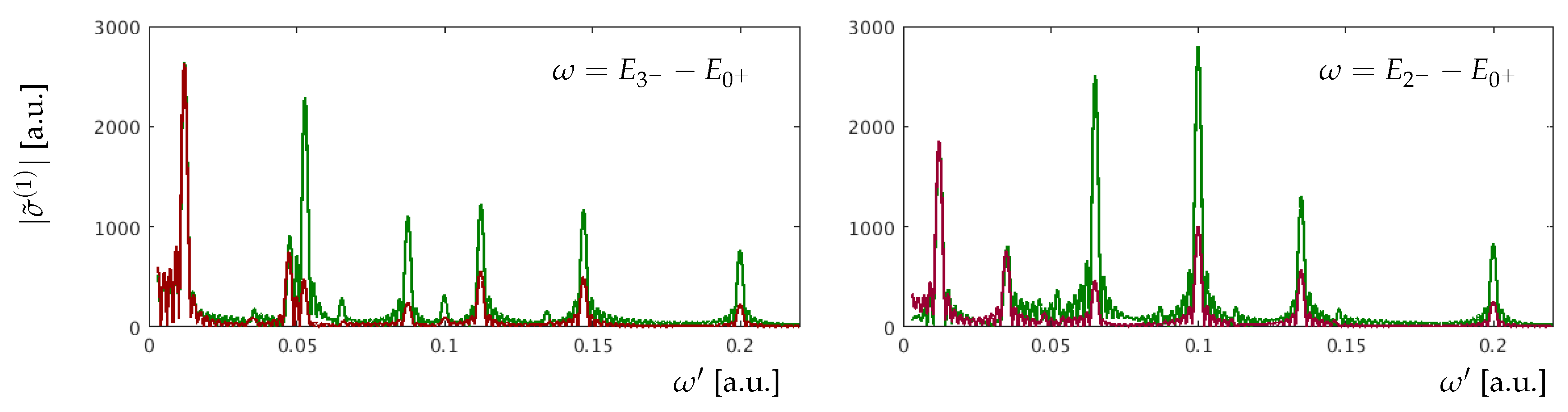
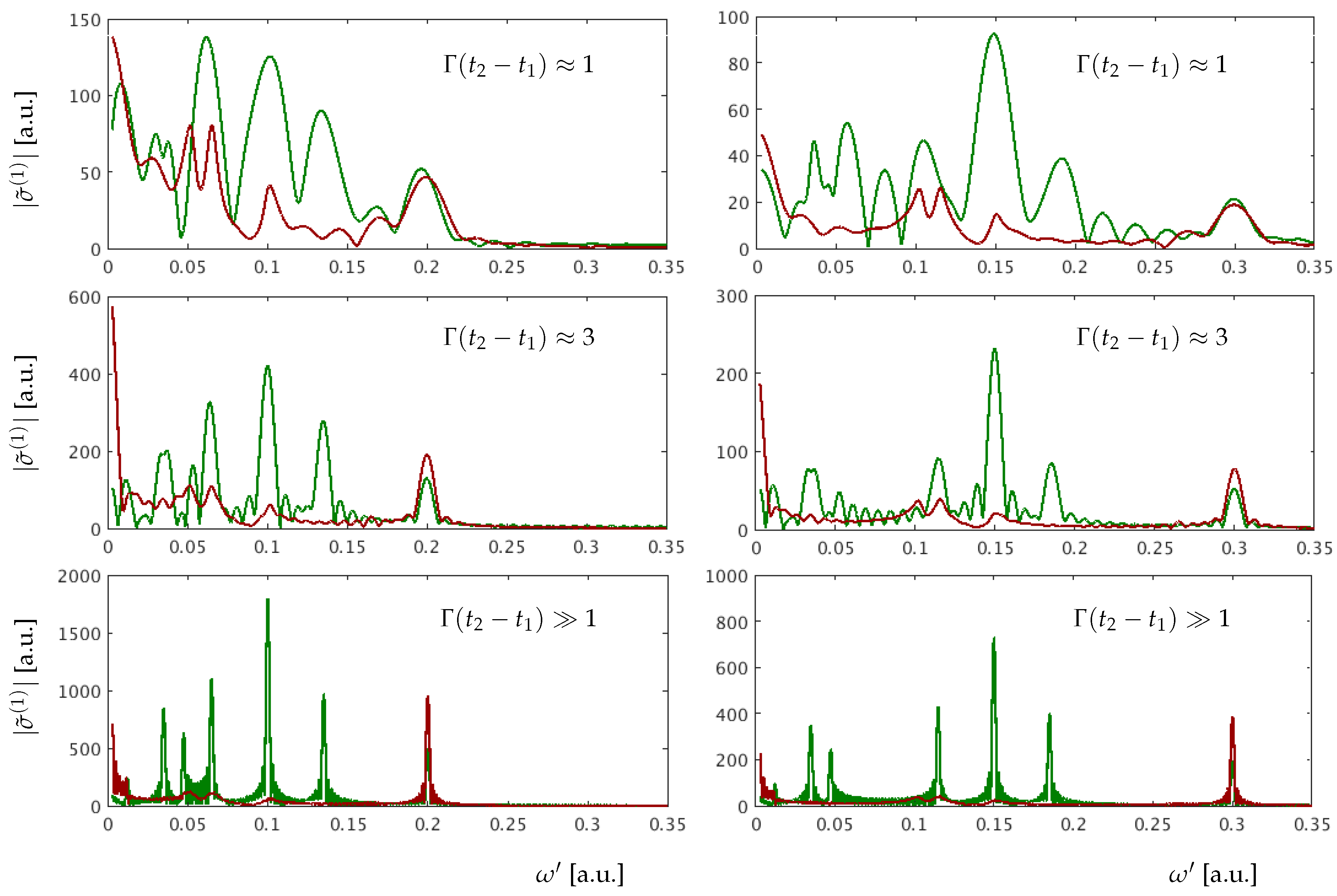
3. Results for a Model Atom
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| XUV | extreme ultraviolet |
| ATA | attosecond transient absorption |
| LIS | light-induced state |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Condition | Expression |
|---|---|---|
| 0 | ||
| 0 | ||
References
- Tamai, N.; Miyasaka, H. Ultrafast dynamics of photochromic systems. Chem. Rev. 2000, 100, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Joly, A.G.; Nelson, K.A. Femtosecond transient absorption spectroscopy of chromium hexacarbonyl in methanol: Observation of initial excited states and carbon monoxide dissociation. J. Phys. Chem. 1989, 93, 2876–2878. [Google Scholar] [CrossRef]
- Pfeifer, T.; Spielmann, C.; Gerber, G. Femtosecond X-ray science. Rep. Prog. Phys. 2006, 69, 443–505. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry. Atomic-scale dynamics of the chemical bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Dhar, L.; Rogers, J.A.; Nelson, K.A. Time-resolved vibrational spectroscopy in the impulsive limit. Chem. Rev. 1994, 94, 157–193. [Google Scholar] [CrossRef]
- Petek, H.; Ogawa, S. Femtosecond time-resolved two-photon photoemission studies of electron dynamics in metals. Prog. Surf. Sci. 1997, 56, 239–310. [Google Scholar] [CrossRef]
- Othonos, A. Probing ultrafast carrier and phonon dynamics in semiconductors. J. Appl. Phys. 1998, 83, 1789–1830. [Google Scholar] [CrossRef]
- Voisin, C.; Fatti, N.D.; Christofilos, D.; Vallee, F. Ultrafast electron dynamics and optical nonlinearities in metal nanoparticles. J. Phys. Chem. B 2001, 105, 2264–2280. [Google Scholar] [CrossRef]
- Stolow, A.; Bragg, A.E.; Neumark, D.M. Femtosecond time-resolved photoelectron spectroscopy. Chem. Rev. 2004, 104, 1719–1758. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski-Tinten, K.; Von Der Linde, D. Ultrafast phase transitions and lattice dynamics probed using laser-produced X-ray pulses. J. Phys. Condens. Matter 2004, 16, R1517–R1536. [Google Scholar] [CrossRef]
- Güdde, J.; Höfer, U. Femtosecond time-resolved studies of image-potential states at surfaces and interfaces of rare-gas adlayers. Prog. Surf. Sci. 2005, 80, 49–91. [Google Scholar] [CrossRef]
- Nibbering, E.T.J.; Fidder, H.; Pines, E. Ultrafast chemistry: Using time-resolved vibrational spectroscopy for interrogation of structural dynamics. Annu. Rev. Phys. Chem. 2005, 56, 337–367. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Gühr, M.; Bargheer, M.; Fushitani, M.; Kiljunen, T.; Schwentner, N. Ultrafast dynamics of halogens in rare gas solids. Phys. Chem. Chem. Phys. 2007, 9, 779–801. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, V. Femtobiology. Annu. Rev. Phys. Chem. 2008, 59, 53–77. [Google Scholar] [CrossRef]
- Southworth, S.H.; Arms, D.A.; Dufresne, E.M.; Dunford, R.W.; Ederer, D.L.; Höhr, C.; Kanter, E.P.; Krässig, B.; Landahl, E.C.; Peterson, E.R.; et al. K-edge X-ray-absorption spectroscopy of laser-generated Kr+ and Kr2+. Phys. Rev. A 2007, 76, 043421. [Google Scholar] [CrossRef]
- Loh, Z.H.; Khalil, M.; Correa, R.E.; Santra, R.; Buth, C.; Leone, S.R. Quantum state-resolved probing of strong-field-ionized xenon atoms using femtosecond high-order harmonic transient absorption spectroscopy. Phys. Rev. Lett. 2007, 98, 143601. [Google Scholar] [CrossRef]
- Young, L.; Arms, D.A.; Dufresne, E.M.; Dunford, R.W.; Ederer, D.L.; Höhr, C.; Kanter, E.P.; Krässig, B.; Landahl, E.C.; Peterson, E.R.; et al. X-ray microprobe of orbital alignment in strong-field ionized atoms. Phys. Rev. Lett. 2006, 97, 083601. [Google Scholar] [CrossRef] [PubMed]
- Santra, R.; Dunford, R.W.; Young, L. Spin-orbit effect on strong-field ionization of krypton. Phys. Rev. A 2006, 74, 043403. [Google Scholar] [CrossRef]
- Johnsson, P.; Mauritsson, J.; Remetter, T.; L’Huillier, A.; Schafer, K.J. Attosecond control of ionization by wave-packet interference. Phys. Rev. Lett. 2007, 99, 233001. [Google Scholar] [CrossRef]
- Glover, T.E.; Hertlein, M.P.; Southworth, S.H.; Allison, T.K.; van Tilborg, J.; Kanter, E.P.; Krässig, B.; Varma, H.R.; Rude, B.; Santra, R.; et al. Controlling X-rays with light. Nat. Phys. 2009, 6, 69–74. [Google Scholar] [CrossRef]
- Ranitovic, P.; Tong, X.M.; Gramkow, B.; De, S.; DePaola, B.; Singh, K.P.; Cao, W.; Magrakvelidze, M.; Ray, D.; Bocharova, I.; et al. IR-assisted ionization of helium by attosecond extreme ultraviolet radiation. New J. Phys. 2010, 12, 013008. [Google Scholar] [CrossRef] [Green Version]
- Wickenhauser, M.; Burgdörfer, J.; Krausz, F.; Drescher, M. Time-resolved Fano resonances. Phys. Rev. Lett. 2005, 94, 023002. [Google Scholar] [CrossRef]
- Buth, C.; Santra, R.; Young, L. Electromagnetically induced transparency for X-rays. Phys. Rev. Lett. 2007, 98, 253001. [Google Scholar] [CrossRef]
- Pfeifer, T.; Abel, M.J.; Nagel, P.M.; Jullien, A.; Loh, Z.-H.; Bell, M.J.; Neumark, D.M.; Leone, S.R. Time-resolved spectroscopy of attosecond quantum dynamics. Chem. Phys. Lett. 2008, 463, 11–24. [Google Scholar] [CrossRef]
- Santra, R.; Buth, C.; Peterson, E.R.; Dunford, R.W.; Kanter, E.P.; Krässig, B.; Southworth, S.H.; Young, L. Strong-field control of X-ray absorption. J. Phys. Conf. Ser. 2007, 88, 012052. [Google Scholar] [CrossRef]
- Buth, C.; Santra, R. Theory of X-ray absorption by laser-dressed atoms. Phys. Rev. A 2007, 75, 033412. [Google Scholar] [CrossRef]
- Buth, C.; Santra, R. X-ray refractive index of laser-dressed atoms. Phys. Rev. A 2008, 78, 043409. [Google Scholar] [CrossRef]
- Porter, G.; Topp, M.R. Nanosecond flash photolysis and the absorption spectra of excited singlet states. Nature 1968, 220, 1228–1229. [Google Scholar] [CrossRef]
- Dantus, M.; Rosker, M.J.; Zewail, A.H. Real-time femtosecond probing of “transition states” in chemical reactions. J. Chem. Phys. 1987, 87, 2395–2397. [Google Scholar] [CrossRef]
- Bressler, C.; Chergui, M. Ultrafast X-ray absorption spectroscopy. Chem. Rev. 2004, 104, 1781–1812. [Google Scholar] [CrossRef]
- Chen, L.X. Probing transient molecular structures in photochemical processes using laser-initiated time-resolved X-ray absorption spectroscopy. Annu. Rev. Phys. Chem. 2005, 56, 221–254. [Google Scholar] [CrossRef]
- Seres, E.; Spielmann, C. Ultrafast soft X-ray absorption spectroscopy with sub-20-fs resolution. Appl. Phys. Lett. 2007, 91, 121919. [Google Scholar] [CrossRef]
- McPherson, A.; Gibson, G.; Jara, H.; Johann, U.; Luk, T.S.; McIntyre, I.A.; Boyer, K.; Rhodes, C.K. Studies of multiphoton production of vacuum-ultraviolet radiation in the rare gases. J. Opt. Soc. Am. B 1987, 4, 595–601. [Google Scholar] [CrossRef]
- Ferray, M.; L’Huillier, A.; Li, X.F.; Lompré, L.A.; Mainfray, G.; Manus, C. Multiple-harmonic conversion of 1064 nm radiation in rare gases. J. Phys. B 1988, 21, L31–L35. [Google Scholar] [CrossRef]
- Corkum, P.B. Plasma perspective on strong field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [Green Version]
- Krause, J.L.; Schafer, K.J.; Kulander, K.C. High-order harmonic generation from atoms and ions in the high intensity regime. Phys. Rev. Lett. 1992, 68, 3535–3538. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.M.; Toma, E.S.; Breger, P.; Mullot, G.; Augé, F.; Balcou, P.; Muller, H.G.; Agostini, P. Observation of a train of attosecond pulses from high harmonic generation. Science 2001, 292, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Goulielmakis, E.; Loh, Z.-H.; Wirth, A.; Santra, R.; Rohringer, N.; Yakovlev, V.S.; Zherebtsov, S.; Pfeifer, T.; Azzeer, A.M.; Kling, M.F.; et al. Real-time observation of valence electron motion. Nature 2010, 466, 739–743. [Google Scholar] [CrossRef]
- Blättermann, A.; Ott, C.; Kaldun, A.; Ding, T.; Pfeifer, T. Two-dimensional spectral interpretation of time-dependent absorption near laser-coupled resonances. J. Phys. B 2014, 47, 124008. [Google Scholar] [CrossRef] [Green Version]
- Santra, R.; Yakovlev, V.S.; Pfeifer, T.; Loh, Z.-H. Theory of attosecond transient absorption spectroscopy of strong-field-generated ions. Phys. Rev. A 2011, 83, 033405. [Google Scholar] [CrossRef]
- Pabst, S.; Sytcheva, A.; Moulet, A.; Wirth, A.; Goulielmakis, E.; Santra, R. Theory of attosecond transient-absorption spectroscopy of krypton for overlapping pump and probe pulses. Phys. Rev. A 2012, 86, 063411. [Google Scholar] [CrossRef]
- Baggesen, J.C.; Lindroth, E.; Madsen, L.B. Theory of attosecond absorption spectroscopy in krypton. Phys. Rev. A 2012, 85, 013415. [Google Scholar] [CrossRef]
- Gallmann, L.; Herrmann, J.; Locher, R.; Sabbar, M.; Ludwig, A.; Lucchini, M.; Keller, U. Resolving intra-atomic electron dynamics with attosecond transient absorption spectroscopy. Mol. Phys. 2013, 111, 2243–2250. [Google Scholar] [CrossRef]
- Cao, W.; Warrick, E.R.; Neumark, D.M.; Leone, S.R. Attosecond transient absorption of argon atoms in the vacuum ultraviolet region: Line energy shifts versus coherent population transfer. New J. Phys. 2016, 18, 013041. [Google Scholar] [CrossRef]
- Wu, M.; Chen, S.; Camp, S.; Schafer, K.J.; Gaarde, M.B. Theory of strong-field attosecond transient absorption. J. Phys. B 2016, 49, 062003. [Google Scholar] [CrossRef] [Green Version]
- Mukamel, S.; Loring, R.F. Nonlinear response function for time-domain and frequency-domain four-wave mixing. J. Opt. Soc. Am. B 1986, 3, 595. [Google Scholar] [CrossRef]
- Marx, C.A.; Harbola, U.; Mukamel, S. Nonlinear optical spectroscopy of single, few, and many molecules: Nonequilibrium Green’s function QED approach. Phys. Rev. A 2008, 77, 022110. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, S. Partially-time-ordered Schwinger-Keldysh loop expansion of coherent nonlinear optical susceptibilities. Phys. Rev. A 2008, 77, 023801. [Google Scholar] [CrossRef]
- Biggs, J.D.; Voll, J.A.; Mukamel, S. Coherent nonlinear optical studies of elementary processes in biological complexes: Diagrammatic techniques based on the wave function versus the density matrix. Philos. Trans. R. Soc. A 2012, 370. [Google Scholar] [CrossRef] [PubMed]
- Keldysh, L.V. Ionization in the field of a strong electromagnetic wave. Sov. Phys. JETP 1965, 20, 1307–1314. [Google Scholar]
- Beck, A.R.; Neumark, D.M.; Leone, S.R. Probing ultrafast dynamics with attosecond transient absorption. Chem. Phys. Lett. 2015, 624, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Bell, M.J.; Beck, A.R.; Mashiko, H.; Wu, M.; Pfeiffer, A.N.; Gaarde, M.B.; Neumark, D.M.; Leone, S.R.; Schafer, K.J. Light-induced states in attosecond transient absorption spectra of laser-dressed helium. Phys. Rev. A 2012, 86, 063408. [Google Scholar] [CrossRef]
- Chen, S.; Wu, M.; Gaarde, M.B.; Schafer, K.J. Quantum interference in attosecond transient absorption of laser-dressed helium atoms. Phys. Rev. A 2013, 87, 033408. [Google Scholar] [CrossRef]
- Paul, H. Interference and ‘which way’ information. Opt. Quantum Electron. 1996, 28, 1111–1127. [Google Scholar] [CrossRef]







| 0 | 2.1888 | 2.2238 | 2.2360 | 2.1889 | 2.2238 |
| 0.25 | 0.33 | 0.33 | |
| 0.42 | 0.33 | 0.33 | |
| 0.33 | 0.33 | 0.33 |
| i = 1 | i = 2 | i = 3 | |
|---|---|---|---|
| 1.9889 | 2.0238 | 2.0361 | |
| 2.1888 | 2.2238 | 2.2360 | |
| 2.3888 | 2.4238 | 2.4360 | |
| 2.089 | 2.124 | ||
| 2.289 | 2.324 |
| i = 1 | i = 2 | i = 3 | |
|---|---|---|---|
| 0.0350 | 0.0122 | 0.0472 | |
| 0.1000 | 0.0650 | 0.0528 | |
| 0.1350 | 0.1000 | 0.0878 | |
| 0.0999 | 0.1350 | 0.1472 | |
| 0.0650 | 0.0999 | 0.1122 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolbasova, D.; Santra, R. Analytical Theory of Attosecond Transient Absorption Spectroscopy of Perturbatively Dressed Systems. Appl. Sci. 2019, 9, 1350. https://doi.org/10.3390/app9071350
Kolbasova D, Santra R. Analytical Theory of Attosecond Transient Absorption Spectroscopy of Perturbatively Dressed Systems. Applied Sciences. 2019; 9(7):1350. https://doi.org/10.3390/app9071350
Chicago/Turabian StyleKolbasova, Daria, and Robin Santra. 2019. "Analytical Theory of Attosecond Transient Absorption Spectroscopy of Perturbatively Dressed Systems" Applied Sciences 9, no. 7: 1350. https://doi.org/10.3390/app9071350
APA StyleKolbasova, D., & Santra, R. (2019). Analytical Theory of Attosecond Transient Absorption Spectroscopy of Perturbatively Dressed Systems. Applied Sciences, 9(7), 1350. https://doi.org/10.3390/app9071350