Recent Advances in Ultrafast Structural Techniques

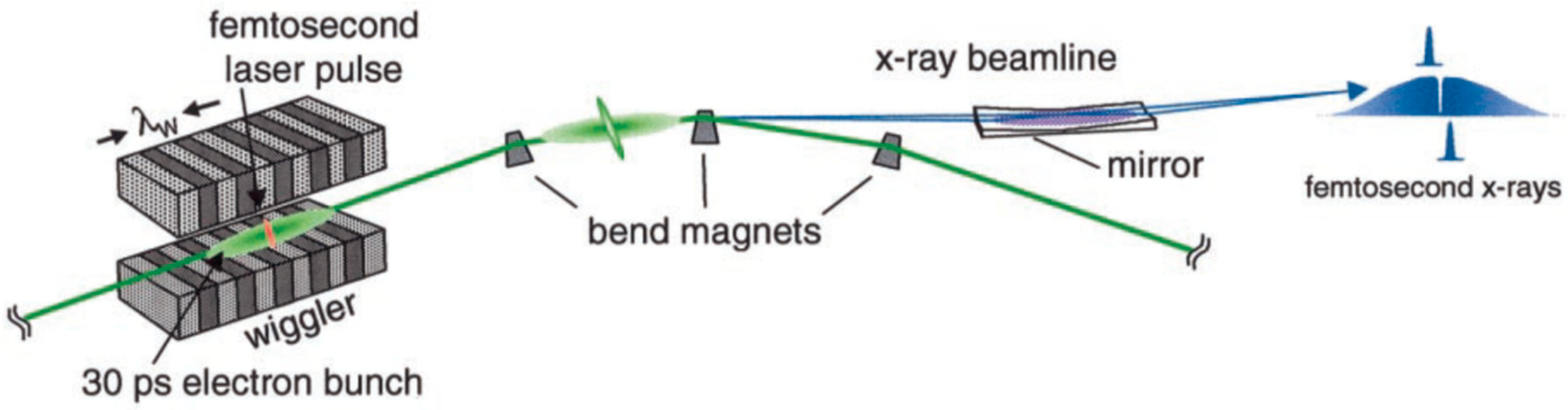{kind=link}
{kind=link}
{kind=link}
{kind=link}
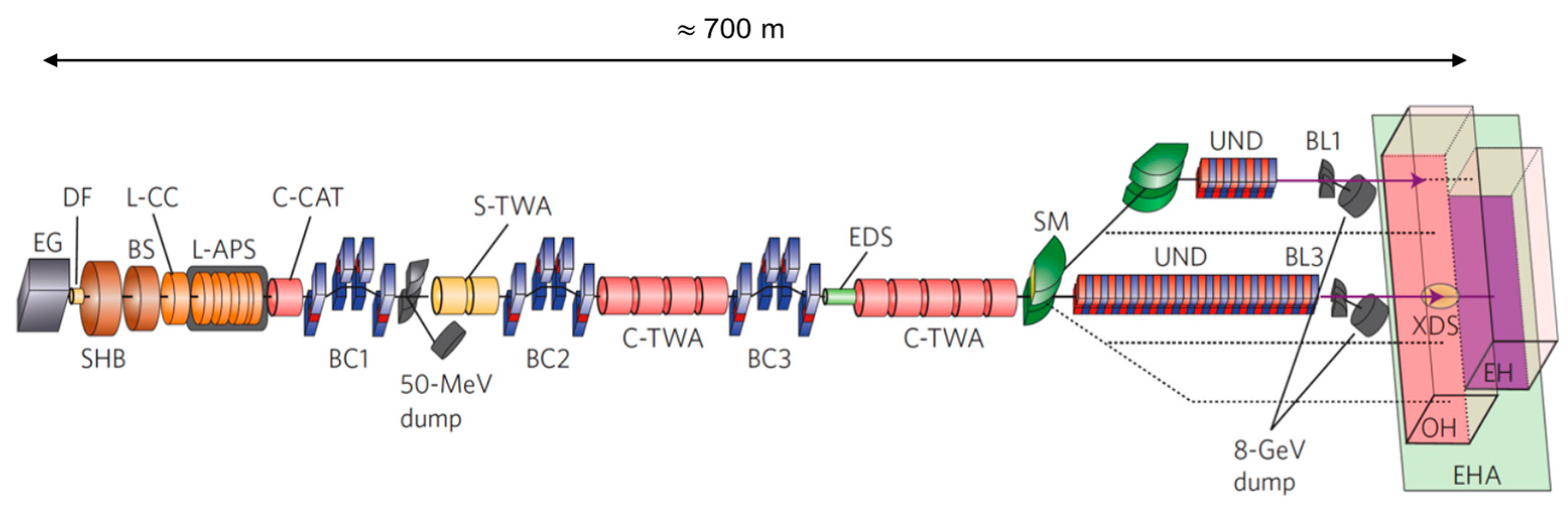{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
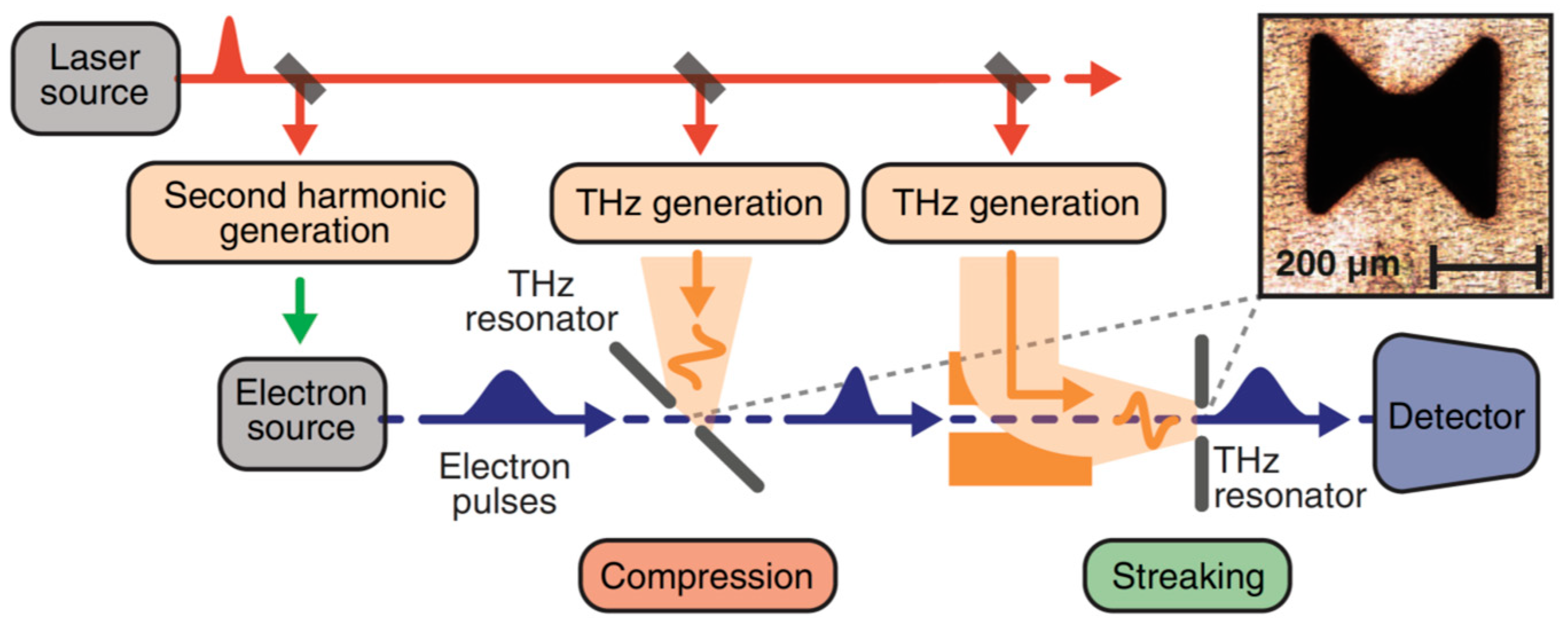{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structural Techniques Involving Ultrashort X-ray Pulses
2.1. Laser Slicing at Third-Generation Synchrotron Facilities
2.2. Table-Top fs Hard X-ray Plasma Diffractometers
2.3. Introduction to Fourth-Generation Fs-Hard X-ray Sources: X-ray Free Electron Lasers
2.4. Main Applications at X-ray Free Electron Lasers (XFELs) for the Study of Structure and Dynamics of Matter
2.4.1. Serial Fs-X-ray Crystallography
2.4.2. Time-Resolved Fs-X-ray Crystallography and Scattering
2.4.3. Fs-X-ray Absorption Spectroscopy
2.5. Timing Techniques at XFEL Facilities
3. Structural Techniques Involving Ultrashort Electron Pulses
3.1. Introduction to Femtosecond Electron Diffraction (FED)
3.2. Electrons as Structural Probes: Fermionic Nature and Source Brightness
3.3. Transverse Electron Beam Properties: Transverse Emittance and Coherence Length
3.4. Temporal Resolution in the Absence of Repulsive Interactions
3.5. Bright Compact Direct Current (DC) FED Setups
3.6. Ultrabright Hybrid DC-Radio Frequency (RF) FED Setups: Compression of Electron Pulses
3.7. Ultrabright Relativistic FED Setups: RF-Photoinjectors
3.8. Compact Ultrafast Low-Energy FED: Metallic Tip as Coherent Electron Sources
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zewail, A.H. 4D Ultrafast Electron Diffraction, Crystallography, and Microscopy. Annu. Rev. Phys. Chem. 2006, 57, 65–103. [Google Scholar] [CrossRef] [PubMed]
- Chergui, M.; Zewail, A.H. Electron and X-ray Methods of Ultrafast Structural Dynamics: Advances and Applications. ChemPhysChem 2009, 10, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Zewail, A.H. Four-Dimensional Electron Microscopy. Science 2010, 328, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Sciaini, G.; Miller, R.J.D. Femtosecond Electron Diffraction: Heralding the Era of Atomically Resolved Dynamics. Rep. Prog. Phys. 2011, 74, 096101. [Google Scholar] [CrossRef]
- Hada, M.; Pichugin, K.; Sciaini, G. Ultrafast Structural Dynamics with Table Top Femtosecond Hard X-ray and Electron Diffraction Setups. Eur. Phys. J. Spec. Top. 2013, 222, 1093–1123. [Google Scholar] [CrossRef]
- Elsaesser, T.; Woerner, M. Perspective: Structural Dynamics in Condensed Matter Mapped by Femtosecond X-ray Diffraction. J. Chem. Phys. 2014, 140, 020901. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.J.D. Femtosecond Crystallography with Ultrabright Electrons and X-rays: Capturing Chemistry in Action. Science 2014, 343, 1108–1116. [Google Scholar] [CrossRef]
- Rumble, J. CRC Handbook of Chemistry and Physics, 99th ed.; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Polanyi, J.C.; Zewail, A.H. Direct Observation of the Transition State. Acc. Chem. Res. 1995, 28, 119–132. [Google Scholar] [CrossRef]
- Grüner, G. The Dynamics of Charge-Density Waves. Rev. Mod. Phys. 1988, 60, 1129–1181. [Google Scholar] [CrossRef]
- Schrieffer, J.R. Theory of Superconductivity; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Allen, P.G.; Mini, S.M.; Perry, D.L.; Stock, S.R. Applications of Synchrotron Radiation Techniques to Materials Science IV: Volume 678, 1st ed.; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- Schotte, F.; Lim, M.; Jackson, T.A.; Smirnov, A.V.; Soman, J.; Olson, J.S.; Phillips, G.N.; Wulff, M.; Anfinrud, P.A. Watching a Protein as It Functions with 150-Ps Time-Resolved X-ray Crystallography. Science 2003, 300, 1944–1947. [Google Scholar] [CrossRef] [PubMed]
- Collet, E.; Lemée-Cailleau, M.-H.; Cointe, M.B.-L.; Cailleau, H.; Wulff, M.; Luty, T.; Koshihara, S.-Y.; Meyer, M.; Toupet, L.; Rabiller, P.; et al. Laser-Induced Ferroelectric Structural Order in an Organic Charge-Transfer Crystal. Science 2003, 300, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Schoenlein, R.W.; Chattopadhyay, S.; Chong, H.H.W.; Glover, T.E.; Heimann, P.A.; Shank, C.V.; Zholents, A.A.; Zolotorev, M.S. Generation of Femtosecond Pulses of Synchrotron Radiation. Science 2000, 287, 2237–2240. [Google Scholar] [CrossRef] [PubMed]
- Zholents, A.A.; Zolotorev, M.S. Femtosecond X-ray Pulses of Synchrotron Radiation. Phys. Rev. Lett. 1996, 76, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Cavalleri, A.; Wall, S.; Simpson, C.; Statz, E.; Ward, D.W.; Nelson, K.A.; Rini, M.; Schoenlein, R.W. Tracking the Motion of Charges in a Terahertz Light Field by Femtosecond X-ray Diffraction. Nature 2006, 442, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Beaud, P.; Johnson, S.L.; Streun, A.; Abela, R.; Abramsohn, D.; Grolimund, D.; Krasniqi, F.; Schmidt, T.; Schlott, V.; Ingold, G. Spatiotemporal Stability of a Femtosecond Hard—X-ray Undulator Source Studied by Control of Coherent Optical Phonons. Phys. Rev. Lett. 2007, 99, 174801. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Beaud, P.; Milne, C.J.; Krasniqi, F.S.; Zijlstra, E.S.; Garcia, M.E.; Kaiser, M.; Grolimund, D.; Abela, R.; Ingold, G. Nanoscale Depth-Resolved Coherent Femtosecond Motion in Laser-Excited Bismuth. Phys. Rev. Lett. 2008, 100, 155501. [Google Scholar] [CrossRef]
- Huber, T.; Mariager, S.O.; Ferrer, A.; Schäfer, H.; Johnson, J.A.; Grübel, S.; Lübcke, A.; Huber, L.; Kubacka, T.; Dornes, C.; et al. Coherent Structural Dynamics of a Prototypical Charge-Density-Wave-to-Metal Transition. Phys. Rev. Lett. 2014, 113, 026401. [Google Scholar] [CrossRef] [PubMed]
- Lantz, G.; Neugebauer, M.J.; Kubli, M.; Savoini, M.; Abreu, E.; Tasca, K.; Dornes, C.; Esposito, V.; Rittmann, J.; Windsor, Y.W.; et al. Coupling between a Charge Density Wave and Magnetism in an Heusler Material. Phys. Rev. Lett. 2017, 119, 227207. [Google Scholar] [CrossRef]
- Porer, M.; Fechner, M.; Bothschafter, E.M.; Rettig, L.; Savoini, M.; Esposito, V.; Rittmann, J.; Kubli, M.; Neugebauer, M.J.; Abreu, E.; et al. Ultrafast Relaxation Dynamics of the Antiferrodistortive Phase in Ca Doped SrTiO3. Phys. Rev. Lett. 2018, 121, 055701. [Google Scholar] [CrossRef]
- Travaglini, G.; Wachter, P.; Marcus, J.; Schlenker, C. The Blue Bronze K0.3MoO3: A New One-Dimensional Conductor. Solid State Commun. 1981, 37, 599–603. [Google Scholar] [CrossRef]
- Martin, I.P.S.; Rehm, G.; Thomas, C.; Bartolini, R. Experience with Low-Alpha Lattices at the Diamond Light Source. Phys. Rev. Spec. Top. Accel. Beams 2011, 14, 040705. [Google Scholar] [CrossRef]
- Murnane, M.M.; Kapteyn, H.C.; Rosen, M.D.; Falcone, R.W. Ultrafast X-ray Pulses from Laser-Produced Plasmas. Science 1991, 251, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Rose-Petruck, C.; Jimenez, R.; Guo, T.; Cavalleri, A.; Siders, C.W.; Rksi, F.; Squier, J.A.; Walker, B.C.; Wilson, K.R.; Barty, C.P.J. Picosecond–Milliångström Lattice Dynamics Measured by Ultrafast X-ray Diffraction. Nature 1999, 398, 310–312. [Google Scholar] [CrossRef]
- Siders, C.W.; Cavalleri, A.; Sokolowski-Tinten, K.; Tóth, C.; Guo, T.; Kammler, M.; von Hoegen, M.H.; Wilson, K.R.; von der Linde, D.; Barty, C.P.J. Detection of Nonthermal Melting by Ultrafast X-ray Diffraction. Science 1999, 286, 1340–1342. [Google Scholar] [CrossRef] [PubMed]
- Cavalleri, A.; Tóth, C.; Siders, C.W.; Squier, J.A.; Ráksi, F.; Forget, P.; Kieffer, J.C. Femtosecond Structural Dynamics in VO2 during an Ultrafast Solid-Solid Phase Transition. Phys. Rev. Lett. 2001, 87, 237401. [Google Scholar] [CrossRef] [PubMed]
- Rousse, A.; Rischel, C.; Fourmaux, S.; Uschmann, I.; Sebban, S.; Grillon, G.; Balcou, P.; Förster, E.; Geindre, J.P.; Audebert, P.; et al. Non-Thermal Melting in Semiconductors Measured at Femtosecond Resolution. Nature 2001, 410, 65–68. [Google Scholar] [CrossRef]
- Sokolowski-Tinten, K.; Blome, C.; Blums, J.; Cavalleri, A.; Dietrich, C.; Tarasevitch, A.; Uschmann, I.; Förster, E.; Kammler, M.; Horn-von-Hoegen, M.; et al. Femtosecond X-ray Measurement of Coherent Lattice Vibrations near the Lindemann Stability Limit. Nature 2003, 422, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, M.; Kutzner, J.; Tsilimis, G.; Zacharias, H. High-Repetition-Rate Hard X-ray Generation with Sub-Millijoule Femtosecond Laser Pulses. Appl. Phys. B 2003, 77, 49–57. [Google Scholar] [CrossRef]
- Bargheer, M.; Zhavoronkov, N.; Gritsai, Y.; Woo, J.C.; Kim, D.S.; Woerner, M.; Elsaesser, T. Coherent Atomic Motions in a Nanostructure Studied by Femtosecond X-ray Diffraction. Science 2004, 306, 1771–1773. [Google Scholar] [CrossRef]
- Serbanescu, C.G.; Chakera, J.A.; Fedosejevs, R. Efficient Kα X-ray Source from Submillijoule Femtosecond Laser Pulses Operated at Kilohertz Repetition Rate. Rev. Sci. Instrum. 2007, 78, 103502. [Google Scholar] [CrossRef]
- Hada, M.; Okimura, K.; Matsuo, J. Characterization of Structural Dynamics of VO2 Thin Film on c-Al2O3 Using in-Air Time-Resolved X-ray Diffraction. Phys. Rev. B 2010, 82, 153401. [Google Scholar] [CrossRef]
- Hada, M.; Okimura, K.; Matsuo, J. Photo-Induced Lattice Softening of Excited-State VO2. Appl. Phys. Lett. 2011, 99, 051903. [Google Scholar] [CrossRef]
- Zamponi, F.; Rothhardt, P.; Stingl, J.; Woerner, M.; Elsaesser, T. Ultrafast Large-Amplitude Relocation of Electronic Charge in Ionic Crystals. Proc. Natl. Acad. Sci. USA 2012, 109, 5207–5212. [Google Scholar] [CrossRef] [PubMed]
- Giles, C.; Celestre, R.; Tasca, K.R.; Dias, C.S.B.; Vescovi, R.; Faria, G.; Ferbonink, G.F.; Nome, R.A. Compact Arrangement for Femtosecond Laser-Induced Generation of Broadband Hard X-ray Pulses. In High-Brightness Sources and Light-Driven Interactions (2018), Paper ET1B.3, Proceedings of the Compact EUV and X-ray Light Sources, Strasbourg, France, 26–28 March 2018; OSA: Washington, DC, USA. [CrossRef]
- Holtz, M.; Hauf, C.; Weisshaupt, J.; Salvador, A.-A.H.; Woerner, M.; Elsaesser, T. Towards Shot-Noise Limited Diffraction Experiments with Table-Top Femtosecond Hard X-ray Sources. Struct. Dyn. 2017, 4, 054304. [Google Scholar] [CrossRef] [PubMed]
- Freyer, B.; Zamponi, F.; Juvé, V.; Stingl, J.; Woerner, M.; Elsaesser, T.; Chergui, M. Ultrafast Inter-Ionic Charge Transfer of Transition-Metal Complexes Mapped by Femtosecond X-ray Powder Diffraction. J. Chem. Phys. 2013, 138, 144504. [Google Scholar] [CrossRef] [PubMed]
- Strickland, D.; Mourou, G. Compression of Amplified Chirped Optical Pulses. Opt. Commun. 1985, 55, 219–221. [Google Scholar] [CrossRef]
- Gao, M.; Lu, C.; Jean-Ruel, H.; Liu, L.C.; Marx, A.; Onda, K.; Koshihara, S.; Nakano, Y.; Shao, X.; Hiramatsu, T.; et al. Mapping Molecular Motions Leading to Charge Delocalization with Ultrabright Electrons. Nature 2013, 496, 343–346. [Google Scholar] [CrossRef]
- Weisshaupt, J.; Juvé, V.; Holtz, M.; Ku, S.; Woerner, M.; Elsaesser, T.; Ališauskas, S.; Pugžlys, A.; Baltuška, A. High-Brightness Table-Top Hard X-ray Source Driven by Sub-100-Femtosecond Mid-Infrared Pulses. Nat. Photonics 2014, 8, 927–930. [Google Scholar] [CrossRef]
- Bentson, L.; Bolton, P.; Bong, E.; Emma, P.; Galayda, J.; Hastings, J.; Krejcik, P.; Rago, C.; Rifkin, J.; Spencer, C.M. FEL Research and Development at the SLAC Sub-Picosecond Photon Source, SPPS. Nucl. Instrum. Methods Phys. Res. A 2003, 507, 205–209. [Google Scholar] [CrossRef]
- Shiner, A.D.; Trallero-Herrero, C.; Kajumba, N.; Bandulet, H.-C.; Comtois, D.; Légaré, F.; Giguère, M.; Kieffer, J.-C.; Corkum, P.B.; Villeneuve, D.M. Wavelength Scaling of High Harmonic Generation Efficiency. Phys. Rev. Lett. 2009, 103, 073902. [Google Scholar] [CrossRef] [PubMed]
- Popmintchev, T.; Chen, M.-C.; Popmintchev, D.; Arpin, P.; Brown, S.; Alisauskas, S.; Andriukaitis, G.; Balciunas, T.; Mucke, O.D.; Pugzlys, A.; et al. Bright Coherent Ultrahigh Harmonics in the KeV X-ray Regime from Mid-Infrared Femtosecond Lasers. Science 2012, 336, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Corkum, P.B. Plasma Perspective on Strong Field Multiphoton Ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Lei, C.; Read, K.; Tobey, R.; Gland, J.; Murnane, M.M.; Kapteyn, H.C. Direct Observation of Surface Chemistry Using Ultrafast Soft-X-ray Pulses. Phys. Rev. Lett. 2001, 87, 025501. [Google Scholar] [CrossRef]
- Stolow, A.; Bragg, A.E.; Neumark, D.M. Femtosecond Time-Resolved Photoelectron Spectroscopy. Chem. Rev. 2004, 104, 1719–1758. [Google Scholar] [CrossRef] [PubMed]
- Itatani, J.; Levesque, J.; Zeidler, D.; Niikura, H.; Pépin, H.; Kieffer, J.C.; Corkum, P.B.; Villeneuve, D.M. Tomographic Imaging of Molecular Orbitals. Nature 2004, 432, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Corkum, P.B.; Krausz, F. Attosecond Science. Nat. Phys. 2007, 3, 381–387. [Google Scholar] [CrossRef]
- Krausz, F.; Ivanov, M. Attosecond Physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Goulielmakis, E.; Loh, Z.-H.; Wirth, A.; Santra, R.; Rohringer, N.; Yakovlev, V.S.; Zherebtsov, S.; Pfeifer, T.; Azzeer, A.M.; Kling, M.F.; et al. Real-Time Observation of Valence Electron Motion. Nature 2010, 466, 739–743. [Google Scholar] [CrossRef]
- Sansone, G.; Kelkensberg, F.; Pérez-Torres, J.F.; Morales, F.; Kling, M.F.; Siu, W.; Ghafur, O.; Johnsson, P.; Swoboda, M.; Benedetti, E.; et al. Electron Localization Following Attosecond Molecular Photoionization. Nature 2010, 465, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Schultze, M.; Fieß, M.; Karpowicz, N.; Gagnon, J.; Korbman, M.; Hofstetter, M.; Neppl, S.; Cavalieri, A.L.; Komninos, Y.; Mercouris, T.; et al. Delay in Photoemission. Science 2010, 328, 1658–1662. [Google Scholar] [CrossRef]
- Torres, R.; Siegel, T.; Brugnera, L.; Procino, I.; Underwood, J.G.; Altucci, C.; Velotta, R.; Springate, E.; Froud, C.; Turcu, I.C.E.; et al. Revealing Molecular Structure and Dynamics through High-Order Harmonic Generation Driven by Mid-IR Fields. Phys. Rev. A 2010, 81, 051802. [Google Scholar] [CrossRef]
- Petersen, J.C.; Kaiser, S.; Dean, N.; Simoncig, A.; Liu, H.Y.; Cavalieri, A.L.; Cacho, C.; Turcu, I.C.E.; Springate, E.; Frassetto, F.; et al. Clocking the Melting Transition of Charge and Lattice Order in 1T-TaS2 with Ultrafast Extreme-Ultraviolet Angle-Resolved Photoemission Spectroscopy. Phys. Rev. Lett. 2011, 107, 177402. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, T.; Hellmann, S.; Wiesenmayer, M.; Sohrt, C.; Stange, A.; Slomski, B.; Carr, A.; Liu, Y.; Avila, L.M.; Kalläne, M.; et al. Collapse of Long-Range Charge Order Tracked by Time-Resolved Photoemission at High Momenta. Nature 2011, 471, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Eich, S.; Stange, A.; Carr, A.V.; Urbancic, J.; Popmintchev, T.; Wiesenmayer, M.; Jansen, K.; Ruffing, A.; Jakobs, S.; Rohwer, T.; et al. Time- and Angle-Resolved Photoemission Spectroscopy with Optimized High-Harmonic Pulses Using Frequency-Doubled Ti:Sapphire Lasers. J. Electron Spectrosc. 2014, 195, 231–236. [Google Scholar] [CrossRef]
- Gariepy, G.; Leach, J.; Kim, K.T.; Hammond, T.J.; Frumker, E.; Boyd, R.W.; Corkum, P.B. Creating High-Harmonic Beams with Controlled Orbital Angular Momentum. Phys. Rev. Lett. 2014, 113, 153901. [Google Scholar] [CrossRef] [PubMed]
- Gessner, O.; Gühr, M. Monitoring Ultrafast Chemical Dynamics by Time-Domain X-ray Photo- and Auger-Electron Spectroscopy. Acc. Chem. Res. 2016, 49, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Marangos, J.P. Development of High Harmonic Generation Spectroscopy of Organic Molecules and Biomolecules. J. Phys. B Atomic Mol. Opt. Phys. 2016, 49, 132001. [Google Scholar] [CrossRef]
- Forbes, R.; Makhija, V.; Veyrinas, K.; Stolow, A.; Lee, J.W.L.; Burt, M.; Brouard, M.; Vallance, C.; Wilkinson, I.; Lausten, R.; et al. Time-Resolved Multi-Mass Ion Imaging: Femtosecond UV-VUV Pump-Probe Spectroscopy with the PImMS Camera. J. Chem. Phys. 2017, 147, 013911. [Google Scholar] [CrossRef]
- Nicholson, C.W.; Lücke, A.; Schmidt, W.G.; Puppin, M.; Rettig, L.; Ernstorfer, R.; Wolf, M. Beyond the Molecular Movie: Dynamics of Bands and Bonds during a Photoinduced Phase Transition. Science 2018, 362, 821–825. [Google Scholar] [CrossRef]
- Young, L.; Ueda, K.; Gühr, M.; Bucksbaum, P.H.; Simon, M.; Mukamel, S.; Rohringer, N.; Prince, K.C.; Masciovecchio, C.; Meyer, M.; et al. Roadmap of Ultrafast X-ray Atomic and Molecular Physics. J. Phys. B Atomic Mol. Opt. Phys. 2018, 51, 032003. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Leone, S.R. Ultrafast X-ray Transient Absorption Spectroscopy of Gas-Phase Photochemical Reactions: A New Universal Probe of Photoinduced Molecular Dynamics. Acc. Chem. Res. 2018, 51, 3203–3211. [Google Scholar] [CrossRef]
- Tanaka, H.; Ishikawa, T.; Aoyagi, H.; Asaka, T.; Asano, Y.; Azumi, N.; Bizen, T.; Ego, H.; Fukami, K.; Fukui, T.; et al. A Compact X-ray Free-Electron Laser Emitting in the Sub-Ångström Region. Nat. Photonics 2012, 6, 540–544. [Google Scholar]
- Doerr, A. The New XFELs. Nat. Methods 2018, 15, 33. [Google Scholar] [CrossRef]
- Abela, R.; Beaud, P.; van Bokhoven, J.A.; Chergui, M.; Feurer, T.; Haase, J.; Ingold, G.; Johnson, S.L.; Knopp, G.; Lemke, H.; et al. Perspective: Opportunities for Ultrafast Science at SwissFEL. Struct. Dyn. 2017, 4, 061602. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.J.; Schietinger, T.; Aiba, M.; Alarcon, A.; Alex, J.; Anghel, A.; Arsov, V.; Beard, C.; Beaud, P.; Bettoni, S.; et al. SwissFEL: The Swiss X-ray Free Electron Laser. Appl. Sci. 2017, 7, 720. [Google Scholar] [CrossRef]
- Arthur, J.; Materlik, G.; Tatchyn, R.; Winick, H. The LCLS: A Fourth Generation Light Source Using the SLAC Linac. Rev. Sci. Instrum. 1995, 66, 1987–1989. [Google Scholar] [CrossRef]
- Cartlidge, E. European XFEL to Shine as Brightest, Fastest X-ray Source. Science 2016, 354, 22–23. [Google Scholar] [CrossRef]
- Helml, W.; Grguraš, I.; Juranić, P.; Düsterer, S.; Mazza, T.; Maier, A.; Hartmann, N.; Ilchen, M.; Hartmann, G.; Patthey, L.; et al. Ultrashort Free-Electron Laser X-ray Pulses. Appl. Sci. 2017, 7, 915. [Google Scholar] [CrossRef]
- Emma, P.; Bane, K.; Cornacchia, M.; Huang, Z.; Schlarb, H.; Stupakov, G.; Walz, D. Femtosecond and Subfemtosecond X-ray Pulses from a Self-Amplified Spontaneous-Emission–Based Free-Electron Laser. Phys. Rev. Lett. 2004, 92, 074801. [Google Scholar] [CrossRef]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; de Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the Full-Length Glucagon Class B G-Protein-Coupled Receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef]
- Ishigami, I.; Zatsepin, N.A.; Hikita, M.; Conrad, C.E.; Nelson, G.; Coe, J.D.; Basu, S.; Grant, T.D.; Seaberg, M.H.; Sierra, R.G.; et al. Crystal Structure of CO-Bound Cytochrome c Oxidase Determined by Serial Femtosecond X-ray Crystallography at Room Temperature. Proc. Natl. Acad. Sci. USA 2017, 114, 8011–8016. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.D.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of Photosystem II and Substrate Binding at Room Temperature. Nature 2016, 540, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Safari, C.; Dods, R.; Nango, E.; Tanaka, R.; Yamashita, A.; Nakane, T.; Tono, K.; Joti, Y.; Båth, P.; et al. Serial Femtosecond Crystallography Structure of Cytochrome c Oxidase at Room Temperature. Sci. Rep. 2017, 7, 4518. [Google Scholar] [CrossRef] [PubMed]
- Ayyer, K.; Yefanov, O.M.; Oberthür, D.; Roy-Chowdhury, S.; Galli, L.; Mariani, V.; Basu, S.; Coe, J.; Conrad, C.E.; Fromme, R.; et al. Macromolecular Diffractive Imaging Using Imperfect Crystals. Nature 2016, 530, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Hanashima, S.; Suzuki, M.; Saiki, H.; Hayashi, T.; Kakinouchi, K.; Sugiyama, S.; Kawatake, S.; Matsuoka, S.; Matsumori, N.; et al. Membrane Protein Structure Determination by SAD, SIR, or SIRAS Phasing in Serial Femtosecond Crystallography Using an Iododetergent. Proc. Natl. Acad. Sci. USA 2016, 113, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Pande, K.; Basu, S.; Tenboer, J. Room Temperature Structures beyond 1.5 Å by Serial Femtosecond Crystallography. Struct. Dyn. 2015, 2, 041708. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial Femtosecond Crystallography of G Protein–Coupled Receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef]
- Redecke, L.; Nass, K.; DePonte, D.P.; White, T.A.; Rehders, D.; Barty, A.; Stellato, F.; Liang, M.; Barends, T.R.M.; Boutet, S.; et al. Natively Inhibited Trypanosoma Brucei Cathepsin B Structure Determined by Using an X-ray Laser. Science 2013, 339, 227–230. [Google Scholar] [CrossRef]
- Johansson, L.C.; Arnlund, D.; White, T.A.; Katona, G.; DePonte, D.P.; Weierstall, U.; Doak, R.B.; Shoeman, R.L.; Lomb, L.; Malmerberg, E.; et al. Lipidic Phase Membrane Protein Serial Femtosecond Crystallography. Nat. Methods 2012, 9, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Fromme, P.; Spence, J.C. Femtosecond Nanocrystallography Using X-ray Lasers for Membrane Protein Structure Determination. Curr. Opin. Struct. Biol. 2011, 21, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray Protein Nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.M.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; Foucar, L.; Botha, S.; Doak, R.B.; Shoeman, R.L.; Nass, K.; Koglin, J.E.; Williams, G.J.; Boutet, S.; Messerschmidt, M.; et al. De Novo Protein Crystal Structure Determination from X-ray Free-Electron Laser Data. Nature 2014, 505, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal Structure of Rhodopsin Bound to Arrestin by Femtosecond X-ray Laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Gati, C.; Laksmono, H.; Dao, E.H.; Gul, S.; Fuller, F.; Kern, J.; Chatterjee, R.; Ibrahim, M.; Brewster, A.S.; et al. Concentric-Flow Electrokinetic Injector Enables Serial Crystallography of Ribosome and Photosystem II. Nat. Methods 2016, 13, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Barty, A.; Caleman, C.; Aquila, A.; Timneanu, N.; Lomb, L.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Self-Terminating Diffraction Gates Femtosecond X-ray Nanocrystallography Measurements. Nat. Photonics 2012, 6, 35–40. [Google Scholar] [CrossRef]
- Scott, H.A. Cretin—A Radiative Transfer Capability for Laboratory Plasmas. J. Quantum Spectrosc. Radiat. Transf. 2001, 71, 689–701. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial Millisecond Crystallography for Routine Room-Temperature Structure Determination at Synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Kunde, T.; Schmidt, B.M. Microcrystal Electron Diffraction (MicroED) for Small-Molecule Structure Determination. Angew. Chem. Int. Ed. 2019, 58, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Martynowycz, M.W.; Gonen, T. From Electron Crystallography of 2D Crystals to MicroED of 3D Crystals. Curr. Opin. Colloid Interface Sci. 2018, 34, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Shinzawa-Itoh, K.; Yano, N.; Takemura, S.; Kato, K.; Hatanaka, M.; Muramoto, K.; Kawahara, T.; Tsukihara, T.; Yamashita, E.; et al. Determination of Damage-Free Crystal Structure of an X-ray–Sensitive Protein Using an XFEL. Nat. Methods 2014, 11, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native Structure of Photosystem II at 1.95 Å Resolution Viewed by Femtosecond X-ray Pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.C.; Mehrabi, P.; Müller-Werkmeister, H.M.; Tellkamp, F.; Jha, A.; Stuart, W.; Persch, E.; Gasparo, R.D.; Diederich, F.; Pai, E.F.; et al. The Hit-and-Return System Enables Efficient Time-Resolved Serial Synchrotron Crystallography. Nat. Methods 2018, 15, 901. [Google Scholar] [CrossRef] [PubMed]
- Zarrine-Afsar, A.; Müller, C.; Talbot, F.O.; Miller, R.J.D. Self-Localizing Stabilized Mega-Pixel Picoliter Arrays with Size-Exclusion Sorting Capabilities. Anal. Chem. 2011, 83, 767–773. [Google Scholar] [CrossRef]
- Zarrine-Afsar, A.; Barends, T.R.M.; Mueller, C.; Fuchs, M.R.; Lomb, L.; Schlichting, I.; Miller, R.J.D. Crystallography on a Chip. Acta Crystallogr. D 2012, 68, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Marx, A.; Epp, S.W.; Zhong, Y.; Kuo, A.; Balo, A.R.; Soman, J.; Schotte, F.; Lemke, H.T.; Owen, R.L.; et al. Fixed Target Matrix for Femtosecond Time-Resolved and in Situ Serial Micro-Crystallography. Struct. Dyn. 2015, 2, 054302. [Google Scholar] [CrossRef]
- Lindenberg, A.M.; Larsson, J.; Sokolowski-Tinten, K.; Gaffney, K.J.; Blome, C.; Synnergren, O.; Sheppard, J.; Caleman, C.; MacPhee, A.G.; Weinstein, D.; et al. Atomic-Scale Visualization of Inertial Dynamics. Science 2005, 308, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.M.; Reis, D.A.; Adams, B.; Akre, R.A.; Arthur, J.; Blome, C.; Bucksbaum, P.H.; Cavalieri, A.L.; Engemann, S.; Fahy, S.; et al. Ultrafast Bond Softening in Bismuth: Mapping a Solid’s Interatomic Potential with X-rays. Science 2007, 315, 633–636. [Google Scholar] [CrossRef]
- Kupitz, C.; Basu, S.; Grotjohann, I.; Fromme, R.; Zatsepin, N.A.; Rendek, K.N.; Hunter, M.S.; Shoeman, R.L.; White, T.A.; Wang, D.; et al. Serial Time-Resolved Crystallography of Photosystem II Using a Femtosecond X-ray Laser. Nature 2014, 513, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Tenboer, J.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-Resolved Serial Crystallography Captures High-Resolution Intermediates of Photoactive Yellow Protein. Science 2014, 346, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; Foucar, L.; Ardevol, A.; Nass, K.; Aquila, A.; Botha, S.; Doak, R.B.; Falahati, K.; Hartmann, E.; Hilpert, M.; et al. Direct Observation of Ultrafast Collective Motions in CO Myoglobin upon Ligand Dissociation. Science 2015, 350, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Pande, K.; Hutchison, C.D.M.; Groenhof, G.; Aquila, A.; Robinson, J.S.; Tenboer, J.; Basu, S.; Boutet, S.; DePonte, D.P.; Liang, M.; et al. Femtosecond Structural Dynamics Drives the Trans/Cis Isomerization in Photoactive Yellow Protein. Science 2016, 352, 725–729. [Google Scholar] [CrossRef]
- Sauter, N.K.; Echols, N.; Adams, P.D.; Zwart, P.H.; Kern, J.; Brewster, A.S.; Koroidov, S.; Alonso-Mori, R.; Zouni, A.; Messinger, J.; et al. No Observable Conformational Changes in PSII. Nature 2016, 533, E1–E2. [Google Scholar] [CrossRef]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A Three-Dimensional Movie of Structural Changes in Bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef]
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A Nanosecond Time-Resolved XFEL Analysis of Structural Changes Associated with CO Release from Cytochrome c Oxidase. Sci. Adv. 2017, 3, e1603042. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore Twisting in the Excited State of a Photoswitchable Fluorescent Protein Captured by Time-Resolved Serial Femtosecond Crystallography. Nat. Chem. 2018, 10, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Arnlund, D.; Johansson, L.C.; Wickstrand, C.; Barty, A.; Williams, G.J.; Malmerberg, E.; Davidsson, J.; Milathianaki, D.; DePonte, D.P.; Shoeman, R.L.; et al. Visualizing a Protein Quake with Time-Resolved X-ray Scattering at a Free-Electron Laser. Nat. Methods 2014, 11, 923–926. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, J.G.; Nozawa, S.; Sato, T.; Oang, K.Y.; Kim, T.W.; Ki, H.; Jo, J.; Park, S.; Song, C.; et al. Direct Observation of Bond Formation in Solution with Femtosecond X-ray Scattering. Nature 2015, 518, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, J.G.; Oang, K.Y.; Kim, T.W.; Ki, H.; Jo, J.; Kim, J.; Sato, T.; Nozawa, S.; Adachi, S.; et al. Femtosecond X-ray Solution Scattering Reveals That Bond Formation Mechanism of a Gold Trimer Complex Is Independent of Excitation Wavelength. Struct. Dyn. 2016, 3, 043209. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.N.; Beitra, L.; Xiong, G.; Higginbotham, A.; Fritz, D.M.; Lemke, H.T.; Zhu, D.; Chollet, M.; Williams, G.J.; Messerschmidt, M.; et al. Ultrafast Three-Dimensional Imaging of Lattice Dynamics in Individual Gold Nanocrystals. Science 2013, 341, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Beaud, P.; Caviezel, A.; Mariager, S.O.; Rettig, L.; Ingold, G.; Dornes, C.; Huang, S.-W.; Johnson, J.A.; Radovic, M.; Huber, T.; et al. A Time-Dependent Order Parameter for Ultrafast Photoinduced Phase Transitions. Nat. Mater. 2014, 13, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Mankowsky, R.; Subedi, A.; Först, M.; Mariager, S.O.; Chollet, M.; Lemke, H.T.; Robinson, J.S.; Glownia, J.M.; Minitti, M.P.; Frano, A.; et al. Nonlinear Lattice Dynamics as a Basis for Enhanced Superconductivity in YBa2Cu3O6.5. Nature 2014, 516, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Lee, W.-S.; Nojiri, H.; Matsuzawa, S.; Yasumura, H.; Nie, L.; Maharaj, A.V.; Gerber, S.; Liu, Y.-J.; Mehta, A.; et al. Ideal Charge-Density-Wave Order in the High-Field State of Superconducting YBCO. Proc. Natl. Acad. Sci. USA 2016, 113, 14645–14650. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, E.; Wittenberg, J.S.; Miller, T.A.; Lutker, K.; Quirin, F.; Lemke, H.; Zhu, D.; Chollet, M.; Robinson, J.; Wen, H.; et al. Visualization of Nanocrystal Breathing Modes at Extreme Strains. Nat. Commun. 2015, 6, 6577. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.; Rettig, L.; Bothschafter, E.M.; Deng, Y.; Dornes, C.; Huber, L.; Huber, T.; Ingold, G.; Inubushi, Y.; Katayama, T.; et al. Dynamics of the Photoinduced Insulator-to-Metal Transition in a Nickelate Film. Struct. Dyn. 2018, 5, 064501. [Google Scholar] [CrossRef]
- Fletcher, L.B.; Lee, H.J.; Döppner, T.; Galtier, E.; Nagler, B.; Heimann, P.; Fortmann, C.; LePape, S.; Ma, T.; Millot, M.; et al. Ultrabright X-ray Laser Scattering for Dynamic Warm Dense Matter Physics. Nat. Photonics 2015, 9, 274–279. [Google Scholar] [CrossRef]
- Gleason, A.E.; Bolme, C.A.; Lee, H.J.; Nagler, B.; Galtier, E.; Milathianaki, D.; Hawreliak, J.; Kraus, R.G.; Eggert, J.H.; Fratanduono, D.E.; et al. Ultrafast Visualization of Crystallization and Grain Growth in Shock-Compressed SiO2. Nat. Commun. 2015, 6, 8191. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.G.; Briggs, R.; McBride, E.E.; Higginbotham, A.; Arnold, B.; Eggert, J.H.; Fratanduono, D.E.; Galtier, E.; Lazicki, A.E.; Lee, H.J.; et al. Direct Observation of Melting in Shock-Compressed Bismuth With Femtosecond X-ray Diffraction. Phys. Rev. Lett. 2015, 115, 095701. [Google Scholar] [CrossRef]
- Goodno, G.D.; Astinov, V.; Miller, R.J.D. Femtosecond Heterodyne-Detected Four-Wave-Mixing Studies of Deterministic Protein Motions. 2. Protein Response. J. Phys. Chem. A 1999, 103, 10630–10643. [Google Scholar] [CrossRef]
- Rosca, F.; Kumar, A.T.N.; Ionascu, D.; Ye, X.; Demidov, A.A.; Sjodin, T.; Wharton, D.; Barrick, D.; Sligar, S.G.; Yonetani, T.; et al. Investigations of Anharmonic Low-Frequency Oscillations in Heme Proteins. J. Phys. Chem. A 2002, 106, 3540–3552. [Google Scholar] [CrossRef]
- Hada, M.; Zhang, D.; Pichugin, K.; Hirscht, J.; Kochman, M.A.; Hayes, S.A.; Manz, S.; Gengler, R.Y.N.; Wann, D.A.; Seki, T.; et al. Cold Ablation Driven by Localized Forces in Alkali Halides. Nat. Commun. 2014, 5, 3863. [Google Scholar] [CrossRef]
- Ihee, H.; Lorenc, M.; Kim, T.K.; Kong, Q.Y.; Cammarata, M.; Lee, J.H.; Bratos, S.; Wulff, M. Ultrafast X-ray Diffraction of Transient Molecular Structures in Solution. Science 2005, 309, 1223–1227. [Google Scholar] [CrossRef]
- Ki, H.; Oang, K.Y.; Kim, J.; Ihee, H. Ultrafast X-ray Crystallography and Liquidography. Annu. Rev. Phys. Chem. 2017, 68, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, T.B.; Kjær, K.S.; Hartsock, R.W.; Dohn, A.O.; Harlang, T.; Chollet, M.; Christensen, M.; Gawelda, W.; Henriksen, N.E.; Kim, J.G.; et al. Atomistic Characterization of the Active-Site Solvation Dynamics of a Model Photocatalyst. Nat. Commun. 2016, 7, 13678. [Google Scholar] [CrossRef] [PubMed]
- Kraus, P.M.; Zürch, M.; Cushing, S.K.; Neumark, D.M.; Leone, S.R. The Ultrafast X-ray Spectroscopic Revolution in Chemical Dynamics. Nat. Rev. Chem. 2018, 2, 82. [Google Scholar] [CrossRef]
- Bressler, C.; Chergui, M. Ultrafast X-ray Absorption Spectroscopy. Chem. Rev. 2004, 104, 1781–1812. [Google Scholar] [CrossRef] [PubMed]
- Bressler, C.; Milne, C.; Pham, V.-T.; ElNahhas, A.; van der Veen, R.M.; Gawelda, W.; Johnson, S.; Beaud, P.; Grolimund, D.; Kaiser, M.; et al. Femtosecond XANES Study of the Light-Induced Spin Crossover Dynamics in an Iron(II) Complex. Science 2009, 323, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.-T.; Penfold, T.J.; van der Veen, R.M.; Lima, F.; El Nahhas, A.; Johnson, S.L.; Beaud, P.; Abela, R.; Bressler, C.; Tavernelli, I.; et al. Probing the Transition from Hydrophilic to Hydrophobic Solvation with Atomic Scale Resolution. J. Am. Chem. Soc. 2011, 133, 12740–12748. [Google Scholar] [CrossRef] [PubMed]
- Huse, N.; Cho, H.; Hong, K.; Jamula, L.; de Groot, F.M.F.; Kim, T.K.; McCusker, J.K.; Schoenlein, R.W. Femtosecond Soft X-ray Spectroscopy of Solvated Transition-Metal Complexes: Deciphering the Interplay of Electronic and Structural Dynamics. J. Phys. Chem. Lett. 2011, 2, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Van Kuiken, B.E.; Cho, H.; Hong, K.; Khalil, M.; Schoenlein, R.W.; Kim, T.K.; Huse, N. Time-Resolved X-ray Spectroscopy in the Water Window: Elucidating Transient Valence Charge Distributions in an Aqueous Fe(II) Complex. J. Phys. Chem. Lett. 2016, 7, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Shelby, M.L.; Lestrange, P.J.; Jackson, N.E.; Haldrup, K.; Mara, M.W.; Stickrath, A.B.; Zhu, D.; Lemke, H.T.; Chollet, M.; Hoffman, B.M.; et al. Ultrafast Excited State Relaxation of a Metalloporphyrin Revealed by Femtosecond X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2016, 138, 8752–8764. [Google Scholar] [CrossRef] [PubMed]
- Mara, M.W.; Hadt, R.G.; Reinhard, M.E.; Kroll, T.; Lim, H.; Hartsock, R.W.; Alonso-Mori, R.; Chollet, M.; Glownia, J.M.; Nelson, S.; et al. Metalloprotein Entatic Control of Ligand-Metal Bonds Quantified by Ultrafast X-ray Spectroscopy. Science 2017, 356, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.A.; Deb, A.; Alonso-Mori, R.; Garabato, B.D.; Glownia, J.M.; Kiefer, L.M.; Koralek, J.; Sikorski, M.; Spears, K.G.; Wiley, T.E.; et al. Polarized XANES Monitors Femtosecond Structural Evolution of Photoexcited Vitamin B12. J. Am. Chem. Soc. 2017, 139, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Gawelda, W.; Pham, V.-T.; Benfatto, M.; Zaushitsyn, Y.; Kaiser, M.; Grolimund, D.; Johnson, S.L.; Abela, R.; Hauser, A.; Bressler, C.; et al. Structural Determination of a Short-Lived Excited Iron(II) Complex by Picosecond X-ray Absorption Spectroscopy. Phys. Rev. Lett. 2007, 98, 057401. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, M.; Bertoni, R.; Lorenc, M.; Cailleau, H.; Di Matteo, S.; Mauriac, C.; Matar, S.F.; Lemke, H.; Chollet, M.; Ravy, S.; et al. Sequential Activation of Molecular Breathing and Bending during Spin-Crossover Photoswitching Revealed by Femtosecond Optical and X-ray Absorption Spectroscopy. Phys. Rev. Lett. 2014, 113, 227402. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, R.; Cammarata, M.; Lorenc, M.; Matar, S.F.; Létard, J.-F.; Lemke, H.T.; Collet, E. Ultrafast Light-Induced Spin-State Trapping Photophysics Investigated in Fe(Phen)2(NCS)2 Spin-Crossover Crystal. Acc. Chem. Res. 2015, 48, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Santomauro, F.G.; Lübcke, A.; Rittmann, J.; Baldini, E.; Ferrer, A.; Silatani, M.; Zimmermann, P.; Grübel, S.; Johnson, J.A.; Mariager, S.O.; et al. Femtosecond X-ray Absorption Study of Electron Localization in Photoexcited Anatase TiO2. Sci. Rep. 2015, 5, 14834. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angela, M.; Anniyev, T.; Beye, M.; Coffee, R.; Föhlisch, A.; Gladh, J.; Katayama, T.; Kaya, S.; Krupin, O.; LaRue, J.; et al. Real-Time Observation of Surface Bond Breaking with an X-ray Laser. Science 2013, 339, 1302–1305. [Google Scholar] [CrossRef]
- Ressler, T. WinXAS: A Program for X-ray Absorption Spectroscopy Data Analysis under MS-Windows. J. Synchrotron Radiat. 1998, 5, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron. Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Cammarata, M.; Feldkamp, J.M.; Fritz, D.M.; Hastings, J.B.; Lee, S.; Lemke, H.T.; Robert, A.; Turner, J.L.; Feng, Y. A Single-Shot Transmissive Spectrometer for Hard X-ray Free Electron Lasers. Appl. Phys. Lett. 2012, 101, 034103. [Google Scholar] [CrossRef]
- Katayama, T.; Inubushi, Y.; Obara, Y.; Sato, T.; Togashi, T.; Tono, K.; Hatsui, T.; Kameshima, T.; Bhattacharya, A.; Ogi, Y.; et al. Femtosecond X-ray Absorption Spectroscopy with Hard X-ray Free Electron Laser. Appl. Phys. Lett. 2013, 103, 131105. [Google Scholar] [CrossRef]
- Grguraš, I.; Maier, A.R.; Behrens, C.; Mazza, T.; Kelly, T.J.; Radcliffe, P.; Düsterer, S.; Kazansky, A.K.; Kabachnik, N.M.; Tschentscher, T.; et al. Ultrafast X-ray Pulse Characterization at Free-Electron Lasers. Nat. Photonics 2012, 6, 852–857. [Google Scholar] [CrossRef]
- Harmand, M.; Coffee, R.; Bionta, M.R.; Chollet, M.; French, D.; Zhu, D.; Fritz, D.M.; Lemke, H.T.; Medvedev, N.; Ziaja, B.; et al. Achieving Few-Femtosecond Time-Sorting at Hard X-ray Free-Electron Lasers. Nat. Photonics 2013, 7, 215–218. [Google Scholar] [CrossRef]
- Cavalieri, A.L.; Fritz, D.M.; Lee, S.H.; Bucksbaum, P.H.; Reis, D.A.; Rudati, J.; Mills, D.M.; Fuoss, P.H.; Stephenson, G.B.; Kao, C.C.; et al. Clocking Femtosecond X Rays. Phys. Rev. Lett. 2005, 94, 114801. [Google Scholar] [CrossRef] [PubMed]
- Bionta, M.R.; Lemke, H.T.; Cryan, J.P.; Glownia, J.M.; Bostedt, C.; Cammarata, M.; Castagna, J.-C.; Ding, Y.; Fritz, D.M.; Fry, A.R.; et al. Spectral Encoding of X-ray/Optical Relative Delay. Opt. Express 2011, 19, 21855–21865. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Decker, F.-J.; Emma, P.; Feng, C.; Field, C.; Frisch, J.; Huang, Z.; Krzywinski, J.; Loos, H.; Welch, J.; et al. Femtosecond X-ray Pulse Characterization in Free-Electron Lasers Using a Cross-Correlation Technique. Phys. Rev. Lett. 2012, 109, 254802. [Google Scholar] [CrossRef]
- Krupin, O.; Trigo, M.; Schlotter, W.F.; Beye, M.; Sorgenfrei, F.; Turner, J.J.; Reis, D.A.; Gerken, N.; Lee, S.; Lee, W.S.; et al. Temporal Cross-Correlation of X-ray Free Electron and Optical Lasers Using Soft X-ray Pulse Induced Transient Reflectivity. Opt. Express 2012, 20, 11396–11406. [Google Scholar] [CrossRef]
- Schorb, S.; Gorkhover, T.; Cryan, J.P.; Glownia, J.M.; Bionta, M.R.; Coffee, R.N.; Erk, B.; Boll, R.; Schmidt, C.; Rolles, D.; et al. X-ray–Optical Cross-Correlator for Gas-Phase Experiments at the Linac Coherent Light Source Free-Electron Laser. Appl. Phys. Lett. 2012, 100, 121107. [Google Scholar] [CrossRef]
- Riedel, R.; Al-Shemmary, A.; Gensch, M.; Golz, T.; Harmand, M.; Medvedev, N.; Prandolini, M.J.; Sokolowski-Tinten, K.; Toleikis, S.; Wegner, U.; et al. Single-Shot Pulse Duration Monitor for Extreme Ultraviolet and X-ray Free-Electron Lasers. Nat. Commun. 2013, 4, 1731. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Togashi, T.; Tono, K.; Inubushi, Y.; Tomizawa, H.; Tanaka, Y.; Adachi, S.; Nakamura, K.; Kodama, R.; Yabashi, M. Development of Ultrafast Pump and Probe Experimental System at SACLA. J. Phys. Conf. Ser. 2013, 425, 092009. [Google Scholar] [CrossRef]
- Hartmann, N.; Helml, W.; Galler, A.; Bionta, M.R.; Grünert, J.; Molodtsov, S.L.; Ferguson, K.R.; Schorb, S.; Swiggers, M.L.; Carron, S.; et al. Sub-Femtosecond Precision Measurement of Relative X-ray Arrival Time for Free-Electron Lasers. Nat. Photonics 2014, 8, 706–709. [Google Scholar] [CrossRef]
- Hebling, J.; Almasi, G.; Kozma, I.; Kuhl, J. Velocity Matching by Pulse Front Tilting for Large Area THz-Pulse Generation. Opt. Express 2002, 10, 1161. [Google Scholar] [CrossRef] [PubMed]
- Hebling, J.; Yeh, K.-L.; Hoffmann, M.C.; Bartal, B.; Nelson, K.A. Generation of High-Power Terahertz Pulses by Tilted-Pulse-Front Excitation and Their Application Possibilities. J. Opt. Soc. Am. B 2008, 25, B6–B19. [Google Scholar] [CrossRef]
- Henke, B.L.; Gullikson, E.M.; Davis, J.C. X-ray Interactions: Photoabsorption, Scattering, Transmission, and Reflection at E = 50–30,000 EV, Z = 1–92. Atomic Data Nucl. Data 1993, 54, 181–342. [Google Scholar] [CrossRef]
- Gahl, C.; Azima, A.; Beye, M.; Deppe, M.; Döbrich, K.; Hasslinger, U.; Hennies, F.; Melnikov, A.; Nagasono, M.; Pietzsch, A.; et al. A Femtosecond X-ray/Optical Cross-Correlator. Nat. Photonics 2008, 2, 165–169. [Google Scholar] [CrossRef]
- Beye, M.; Krupin, O.; Hays, G.; Reid, A.H.; Rupp, D.; de Jong, S.; Lee, S.; Lee, W.-S.; Chuang, Y.-D.; Coffee, R.; et al. X-ray Pulse Preserving Single-Shot Optical Cross-Correlation Method for Improved Experimental Temporal Resolution. Appl. Phys. Lett. 2012, 100, 121108. [Google Scholar] [CrossRef]
- Ischenko, A.A.; Golubkov, V.V.; Spiridonov, V.P.; Zgurskii, A.V.; Akhmanov, A.S.; Vabischevich, M.G.; Bagratashvili, V.N. A Stroboscopical Gas-Electron Diffraction Method for the Investigation of Short-Lived Molecular Species. Appl. Phys. B 1983, 32, 161–163. [Google Scholar] [CrossRef]
- Williamson, S.; Mourou, G.; Li, J.C.M. Time-Resolved Laser-Induced Phase Transformation in Aluminum. Phys. Rev. Lett. 1984, 52, 2364–2367. [Google Scholar] [CrossRef]
- Williamson, J.C.; Dantus, M.; Kim, S.B.; Zewail, A.H. Ultrafast Diffraction and Molecular Structure. Chem. Phys. Lett. 1992, 196, 529–534. [Google Scholar] [CrossRef]
- Dantus, M.; Kim, S.B.; Williamson, J.C.; Zewail, A.H. Ultrafast Electron Diffraction. 5. Experimental Time Resolution and Applications. J. Phys. Chem. 1994, 98, 2782–2796. [Google Scholar] [CrossRef]
- Williamson, J.C.; Cao, J.; Ihee, H.; Frey, H.; Zewail, A.H. Clocking Transient Chemical Changes by Ultrafast Electron Diffraction. Nature 1997, 386, 159–162. [Google Scholar] [CrossRef]
- Ihee, H.; Lobastov, V.A.; Gomez, U.M.; Goodson, B.M.; Srinivasan, R.; Ruan, C.-Y.; Zewail, A.H. Direct Imaging of Transient Molecular Structures with Ultrafast Diffraction. Science 2001, 291, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Dudek, R.C.; Weber, P.M. Ultrafast Diffraction Imaging of the Electrocyclic Ring-Opening Reaction of 1,3-Cyclohexadiene. J. Phys. Chem. A 2001, 105, 4167–4171. [Google Scholar] [CrossRef]
- Yang, J. Diffractive Imaging of Coherent Nuclear Motion in Isolated Molecules. Phys. Rev. Lett. 2016, 117, 153002. [Google Scholar] [CrossRef]
- Yang, J.; Guehr, M.; Vecchione, T.; Robinson, M.S.; Li, R.; Hartmann, N.; Shen, X.; Coffee, R.; Corbett, J.; Fry, A.; et al. Diffractive Imaging of a Rotational Wavepacket in Nitrogen Molecules with Femtosecond Megaelectronvolt Electron Pulses. Nat. Commun. 2016, 7, 11232. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, X.; Wolf, T.J.A.; Li, Z.; Nunes, J.P.F.; Coffee, R.; Cryan, J.P.; Gühr, M.; Hegazy, K.; Heinz, T.F.; et al. Imaging CF3I Conical Intersection and Photodissociation Dynamics with Ultrafast Electron Diffraction. Science 2018, 361, 64–67. [Google Scholar] [CrossRef]
- Ruan, C.-Y.; Lobastov, V.A.; Vigliotti, F.; Chen, S.; Zewail, A.H. Ultrafast Electron Crystallography of Interfacial Water. Science 2004, 304, 80–84. [Google Scholar] [CrossRef]
- Gedik, N.; Yang, D.-S.; Logvenov, G.; Bozovic, I.; Zewail, A.H. Nonequilibrium Phase Transitions in Cuprates Observed by Ultrafast Electron Crystallography. Science 2007, 316, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Baum, P.; Zewail, A.H. Breaking Resolution Limits in Ultrafast Electron Diffraction and Microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 16105–16110. [Google Scholar] [CrossRef] [PubMed]
- Baum, P.; Yang, D.-S.; Zewail, A.H. 4D Visualization of Transitional Structures in Phase Transformations by Electron Diffraction. Science 2007, 318, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Frigge, T.; Hafke, B.; Witte, T.; Krenzer, B.; Streubühr, C.; Samad Syed, A.; Mikšić Trontl, V.; Avigo, I.; Zhou, P.; Ligges, M.; et al. Optically Excited Structural Transition in Atomic Wires on Surfaces at the Quantum Limit. Nature 2017, 544, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Siwick, B.J.; Dwyer, J.R.; Jordan, R.E.; Miller, R.J.D. An Atomic-Level View of Melting Using Femtosecond Electron Diffraction. Science 2003, 302, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Harb, M.; Ernstorfer, R.; Hebeisen, C.T.; Sciaini, G.; Peng, W.; Dartigalongue, T.; Eriksson, M.A.; Lagally, M.G.; Kruglik, S.G.; Miller, R.J.D. Electronically Driven Structure Changes of Si Captured by Femtosecond Electron Diffraction. Phys. Rev. Lett. 2008, 100, 155504. [Google Scholar] [CrossRef] [PubMed]
- Ernstorfer, R.; Harb, M.; Hebeisen, C.T.; Sciaini, G.; Dartigalongue, T.; Miller, R.J.D. The Formation of Warm Dense Matter: Experimental Evidence for Electronic Bond Hardening in Gold. Science 2009, 323, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Sciaini, G.; Harb, M.; Kruglik, S.G.; Payer, T.; Hebeisen, C.T.; zu Heringdorf, F.-J.M.; Yamaguchi, M.; Hoegen, M.H.; Ernstorfer, R.; Miller, R.J.D. Electronic Acceleration of Atomic Motions and Disordering in Bismuth. Nature 2009, 458, 56–59. [Google Scholar] [CrossRef]
- Eichberger, M.; Schäfer, H.; Krumova, M.; Beyer, M.; Demsar, J.; Berger, H.; Moriena, G.; Sciaini, G.; Miller, R.J.D. Snapshots of Cooperative Atomic Motions in the Optical Suppression of Charge Density Waves. Nature 2010, 468, 799–802. [Google Scholar] [CrossRef]
- Musumeci, P.; Moody, J.T.; Scoby, C.M.; Gutierrez, M.S.; Westfall, M. Laser-Induced Melting of a Single Crystal Gold Sample by Time-Resolved Ultrafast Relativistic Electron Diffraction. Appl. Phys. Lett. 2010, 97, 063502. [Google Scholar] [CrossRef]
- Erasmus, N.; Eichberger, M.; Haupt, K.; Boshoff, I.; Kassier, G.; Birmurske, R.; Berger, H.; Demsar, J.; Schwoerer, H. Ultrafast Dynamics of Charge Density Waves in 4Hb TaSe2 Probed by Femtosecond Electron Diffraction. Phys. Rev. Lett. 2012, 109, 167402. [Google Scholar] [CrossRef] [PubMed]
- Eichberger, M.; Erasmus, N.; Haupt, K.; Kassier, G.; von Flotow, A.; Demsar, J.; Schwoerer, H. Femtosecond Streaking of Electron Diffraction Patterns to Study Structural Dynamics in Crystalline Matter. Appl. Phys. Lett. 2013, 102, 121106. [Google Scholar] [CrossRef]
- Daraszewicz, S.L.; Giret, Y.; Naruse, N.; Murooka, Y.; Yang, J.; Duffy, D.M.; Shluger, A.L.; Tanimura, K. Structural Dynamics of Laser-Irradiated Gold Nanofilms. Phys. Rev. B 2013, 88, 184101. [Google Scholar] [CrossRef]
- Chatelain, R.P.; Morrison, V.R.; Klarenaar, B.L.M.; Siwick, B.J. Coherent and Incoherent Electron-Phonon Coupling in Graphite Observed with Radio-Frequency Compressed Ultrafast Electron Diffraction. Phys. Rev. Lett. 2014, 113, 235502. [Google Scholar] [CrossRef] [PubMed]
- Morrison, V.R.; Chatelain, R.P.; Tiwari, K.L.; Hendaoui, A.; Bruhács, A.; Chaker, M.; Siwick, B.J. A Photoinduced Metal-like Phase of Monoclinic VO2 Revealed by Ultrafast Electron Diffraction. Science 2014, 346, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Waldecker, L.; Miller, T.A.; Rudé, M.; Bertoni, R.; Osmond, J.; Pruneri, V.; Simpson, R.E.; Ernstorfer, R.; Wall, S. Time-Domain Separation of Optical Properties from Structural Transitions in Resonantly Bonded Materials. Nat. Mater. 2015, 14, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.F.; Latychevskaia, T.; Pennacchio, F.; Reguera, J.; Stellacci, F.; Carbone, F. Order/Disorder Dynamics in a Dodecanethiol-Capped Gold Nanoparticles Supracrystal by Small-Angle Ultrafast Electron Diffraction. Nano Lett. 2016, 16, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, W.-G.; Wu, L.; Zhu, P.; Konstantinova, T.; Tao, J.; Yang, J.; Cheong, S.-W.; Carbone, F.; Misewich, J.A.; et al. Dichotomy in Ultrafast Atomic Dynamics as Direct Evidence of Polaron Formation in Manganites. npj Quantum Mater. 2016, 1, 16026. [Google Scholar] [CrossRef]
- Wu, X.; Tan, L.Z.; Shen, X.; Hu, T.; Miyata, K.; Trinh, M.T.; Li, R.; Coffee, R.; Liu, S.; Egger, D.A.; et al. Light-Induced Picosecond Rotational Disordering of the Inorganic Sublattice in Hybrid Perovskites. Sci. Adv. 2017, 3, e1602388. [Google Scholar] [CrossRef]
- Lin, M.-F.; Kochat, V.; Krishnamoorthy, A.; Bassman, L.; Weninger, C.; Zheng, Q.; Zhang, X.; Apte, A.; Tiwary, C.S.; Shen, X.; et al. Ultrafast Non-Radiative Dynamics of Atomically Thin MoSe2. Nat. Commun. 2017, 8, 1745. [Google Scholar] [CrossRef]
- Konstantinova, T.; Rameau, J.D.; Reid, A.H.; Abdurazakov, O.; Wu, L.; Li, R.; Shen, X.; Gu, G.; Huang, Y.; Rettig, L.; et al. Nonequilibrium Electron and Lattice Dynamics of Strongly Correlated Bi2Sr2CaCu2O8+δ Single Crystals. Sci. Adv. 2018, 4, eaap7427. [Google Scholar] [CrossRef] [PubMed]
- Sie, E.J.; Nyby, C.M.; Pemmaraju, C.D.; Park, S.J.; Shen, X.; Yang, J.; Hoffmann, M.C.; Ofori-Okai, B.K.; Li, R.; Reid, A.H.; et al. An Ultrafast Symmetry Switch in a Weyl Semimetal. Nature 2019, 565, 61. [Google Scholar] [CrossRef] [PubMed]
- Von Borries, B.; Ruska, E. Versuche, Rechnungen Und Ergebnisse Zur Frage Des Autflösungsvermögens Beim Übermikroskop. Z. Tech. Phys. 1939, 20, 225–235. [Google Scholar]
- Fink, J.H.; Schumacher, B.W. Characterization of Charged Particle Beam Sources. Nucl. Instrum. Methods 1975, 130, 353–358. [Google Scholar] [CrossRef]
- Callaham, M.B. Quantum-Mechanical Constraints on Electron-Beam Brightness. IEEE J. Quantum Electron. 1988, 24, 1958–1962. [Google Scholar] [CrossRef]
- Spence, J.C.H.; Howells, M.R. Synchrotron Soft X-ray and Field-Emission Electron Sources: A Comparison. Ultramicroscopy 2002, 93, 213–222. [Google Scholar] [CrossRef]
- Floettmann, K. Some Basic Features of the Beam Emittance. Phys. Rev. Spec. Top. Acc. Beams 2003, 6, 034202. [Google Scholar] [CrossRef]
- Luiten, O.J.; Claessens, B.J.; Van Der Geer, S.B.; Reijnders, M.P.; Taban, G.; Vredenbregt, E.J.D. Ultracold Electron Sources. Int. J. Mod. Phys. A 2007, 22, 3882–3897. [Google Scholar] [CrossRef]
- Zolotorev, M.; Commins, E.D.; Sannibale, F. Proposal for a Quantum-Degenerate Electron Source. Phys. Rev. Lett. 2007, 98, 184801. [Google Scholar] [CrossRef]
- Bazarov, I.V.; Dunham, B.M.; Sinclair, C.K. Maximum Achievable Beam Brightness from Photoinjectors. Phys. Rev. Lett. 2009, 102, 104801. [Google Scholar] [CrossRef]
- Reed, B.W.; Armstrong, M.R.; Browning, N.D.; Campbell, G.H.; Evans, J.E.; LaGrange, T.; Masiel, D.J. The Evolution of Ultrafast Electron Microscope Instrumentation. Microsc. Microanal. 2009, 15, 272–281. [Google Scholar] [CrossRef]
- Jarvis, J.D.; Andrews, H.L.; Ivanov, B.; Stewart, C.L.; de Jonge, N.; Heeres, E.C.; Kang, W.-P.; Wong, Y.-M.; Davidson, J.L.; Brau, C.A. Resonant Tunneling and Extreme Brightness from Diamond Field Emitters and Carbon Nanotubes. J. Appl. Phys. 2010, 108, 094322. [Google Scholar] [CrossRef]
- Musumeci, P.; Li, R.K. High Brightness Electron Sources for MeV Ultrafast Diffraction and Microscopy. In Ultrafast Nonlinear Imaging and Spectroscopy II; International Society for Optics and Photonics: Bellingham, WA, USA, 2014; Volume 9198, p. 91980S. [Google Scholar] [CrossRef]
- Leemann, S.C.; Streun, A.; Wrulich, A.F. Beam Characterization for the Field-Emitter-Array Cathode-Based Low-Emittance Gun. Phys. Rev. Spec. Top. Accel. Beams 2007, 10, 071302. [Google Scholar] [CrossRef]
- Henderson, R. The Potential and Limitations of Neutrons, Electrons and X-rays for Atomic Resolution Microscopy of Unstained Biological Molecules. Q. Rev. Biophys. 1995, 28, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.R.; Hebeisen, C.T.; Ernstorfer, R.; Harb, M.; Deyirmenjian, V.B.; Jordan, R.E.; Miller, R.J.D. Femtosecond Electron Diffraction: ‘Making the Molecular Movie’. Philos. Trans. R. Soc. A 2006, 364, 741–778. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.J.D.; Ernstorfer, R.; Harb, M.; Gao, M.; Hebeisen, C.T.; Jean-Ruel, H.; Lu, C.; Moriena, G.; Sciaini, G. ‘Making the Molecular Movie’: First Frames. Acta Crystallogr. A 2010, 66, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Baum, P.; Rudolf, P.; Zewail, A.H. Structural Preablation Dynamics of Graphite Observed by Ultrafast Electron Crystallography. Phys. Rev. Lett. 2008, 100, 035501. [Google Scholar] [CrossRef] [PubMed]
- Lahme, S.; Kealhofer, C.; Krausz, F.; Baum, P. Femtosecond Single-Electron Diffraction. Struct. Dyn. 2014, 1, 034303. [Google Scholar] [CrossRef]
- Gahlmann, A.; Tae Park, S.; Zewail, A.H. Ultrashort Electron Pulses for Diffraction, Crystallography and Microscopy: Theoretical and Experimental Resolutions. Phys. Chem. Chem. Phys. 2008, 10, 2894. [Google Scholar] [CrossRef]
- Aidelsburger, M.; Kirchner, F.O.; Krausz, F.; Baum, P. Single-Electron Pulses for Ultrafast Diffraction. Proc. Natl. Acad. Sci. USA 2010, 107, 19714–19719. [Google Scholar] [CrossRef]
- Siwick, B.J.; Dwyer, J.R.; Jordan, R.E.; Miller, R.J.D. Ultrafast Electron Optics: Propagation Dynamics of Femtosecond Electron Packets. J. Appl. Phys. 2002, 92, 1643–1648. [Google Scholar] [CrossRef]
- Reed, B.W. Femtosecond Electron Pulse Propagation for Ultrafast Electron Diffraction. J. Appl. Phys. 2006, 100, 034916. [Google Scholar] [CrossRef]
- Michalik, A.M.; Sipe, J.E. Analytic Model of Electron Pulse Propagation in Ultrafast Electron Diffraction Experiments. J. Appl. Phys. 2006, 99, 054908. [Google Scholar] [CrossRef]
- Michalik, A.M.; Sipe, J.E. Evolution of Non-Gaussian Electron Bunches in Ultrafast Electron Diffraction Experiments: Comparison to Analytic Model. J. Appl. Phys. 2009, 105, 084913. [Google Scholar] [CrossRef]
- General Particle Tracer (GPT) Package Software. Available online: http://www.pulsar.nl/gpt/ (accessed on 1 February 2019).
- ASTRA. A Space Charge Tracking Algorithm. Available online: http://www.desy.de/~mpyflo/ (accessed on 1 February 2019).
- Han, T.-R.T.; Zhou, F.; Malliakas, C.D.; Duxbury, P.M.; Mahanti, S.D.; Kanatzidis, M.G.; Ruan, C.-Y. Exploration of Metastability and Hidden Phases in Correlated Electron Crystals Visualized by Femtosecond Optical Doping and Electron Crystallography. Sci. Adv. 2015, 1, e1400173. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Hayes, S.A.; Keskin, S.; Corthey, G.; Hada, M.; Pichugin, K.; Marx, A.; Hirscht, J.; Shionuma, K.; Onda, K.; et al. Direct Observation of Collective Modes Coupled to Molecular Orbital–Driven Charge Transfer. Science 2015, 350, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Petruk, A.; Pichugin, K.; Sciaini, G. Shaped Cathodes for the Production of Ultra-Short Multi-Electron Pulses. Struct. Dyn. 2017, 4, 044005. [Google Scholar] [CrossRef]
- Korobkin, V.V.; Stepanov, B.M.; Fanchenko, S.D.; Schelev, M.Y. Pico-Femtosecond Electron Optical Photography. Opt. Quantum Electron. 1978, 10, 367–381. [Google Scholar] [CrossRef]
- Poisson Superfish. Available online: http://laacg.lanl.gov/laacg/services/serv_codes.phtml (accessed on 1 February 2019).
- Alpert, D. Initiation of Electrical Breakdown in Ultrahigh Vacuum. J. Vac. Sci. Technol. 1964, 1, 35–50. [Google Scholar] [CrossRef]
- Kildemo, M.; Calatroni, S.; Taborelli, M. Breakdown and Field Emission Conditioning of Cu, Mo, and W. Phys. Rev. Spec. Top. Acc. Beams 2004, 7, 092003. [Google Scholar] [CrossRef]
- Slade, P. The Vacuum Interrupter: Theory, Design, and Application, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Weber, P.M.; Carpenter, S.D.; Lucza, T. Reflectron Design for Femtosecond Electron Guns. In Proceedings of the SPIE 2521, Time-Resolved Electron and X-ray Diffraction, San Diego, CA, USA, 1 September 1995. [Google Scholar]
- Kassier, G.H.; Haupt, K.; Erasmus, N.; Rohwer, E.G.; Schwoerer, H. Achromatic Reflectron Compressor Design for Bright Pulses in Femtosecond Electron Diffraction. J. Appl. Phys. 2009, 105, 113111. [Google Scholar] [CrossRef]
- Tokita, S.; Hashida, M.; Inoue, S.; Nishoji, T.; Otani, K.; Sakabe, S. Single-Shot Femtosecond Electron Diffraction with Laser-Accelerated Electrons: Experimental Demonstration of Electron Pulse Compression. Phys. Rev. Lett. 2010, 105, 215004. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gedik, N. Electron Pulse Compression With a Practical Reflectron Design for Ultrafast Electron Diffraction. IEEE J. Sel. Top. Quantum Electron. 2012, 18, 140–147. [Google Scholar] [CrossRef]
- Mankos, M.; Shadman, K.; Siwick, B.J. A Novel Electron Mirror Pulse Compressor. Ultramicroscopy 2017, 183, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Veisz, L.; Kurkin, G.; Chernov, K.; Tarnetsky, V.; Apolonski, A.; Krausz, F.; Fill, E. Hybrid Dc–Ac Electron Gun for Fs-Electron Pulse Generation. New J. Phys. 2007, 9, 451. [Google Scholar] [CrossRef]
- Van Oudheusden, T.; de Jong, E.F.; van der Geer, S.B.; Op’t Root, W.P.E.M.; Luiten, O.J.; Siwick, B.J. Electron Source Concept for Single-Shot Sub-100 Fs Electron Diffraction in the 100 KeV Range. J. Appl. Phys. 2007, 102, 093501. [Google Scholar] [CrossRef]
- Van Oudheusden, T.; Pasmans, P.L.E.M.; van der Geer, S.B.; de Loos, M.J.; van der Wiel, M.J.; Luiten, O.J. Compression of Subrelativistic Space-Charge-Dominated Electron Bunches for Single-Shot Femtosecond Electron Diffraction. Phys. Rev. Lett. 2010, 105, 264801. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, R.P.; Morrison, V.R.; Godbout, C.; Siwick, B.J. Ultrafast Electron Diffraction with Radio-Frequency Compressed Electron Pulses. Appl. Phys. Lett. 2012, 101, 081901. [Google Scholar] [CrossRef]
- Gao, M.; Jean-Ruel, H.; Cooney, R.R.; Stampe, J.; de Jong, M.; Harb, M.; Sciaini, G.; Moriena, G.; Dwayne Miller, R.J. Full Characterization of RF Compressed Femtosecond Electron Pulses Using Ponderomotive Scattering. Opt. Express 2012, 20, 12048. [Google Scholar] [CrossRef]
- Chollet, M.; Guerin, L.; Uchida, N.; Fukaya, S.; Shimoda, H.; Ishikawa, T.; Matsuda, K.; Hasegawa, T.; Ota, A.; Yamochi, H.; et al. Gigantic Photoresponse in ¼-Filled-Band Organic Salt (EDO-TTF)2PF6. Science 2005, 307, 86–89. [Google Scholar] [CrossRef]
- Onda, K.; Ogihara, S.; Yonemitsu, K.; Maeshima, N.; Ishikawa, T.; Okimoto, Y.; Shao, X.; Nakano, Y.; Yamochi, H.; Saito, G.; et al. Photoinduced Change in the Charge Order Pattern in the Quarter-Filled Organic Conductor (EDO-TTF)2PF6 with a Strong Electron-Phonon Interaction. Phys. Rev. Lett. 2008, 101, 067403. [Google Scholar] [CrossRef] [PubMed]
- Gliserin, A.; Walbran, M.; Baum, P. Passive Optical Enhancement of Laser-Microwave Synchronization. Appl. Phys. Lett. 2013, 103, 031113. [Google Scholar] [CrossRef]
- Gliserin, A.; Walbran, M.; Krausz, F.; Baum, P. Sub-Phonon-Period Compression of Electron Pulses for Atomic Diffraction. Nat. Commun. 2015, 6, 8723. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.R.; René de Cotret, L.P.; Stern, M.J.; Siwick, B.J. Solving the Jitter Problem in Microwave Compressed Ultrafast Electron Diffraction Instruments: Robust Sub-50 Fs Cavity-Laser Phase Stabilization. Struct. Dyn. 2017, 4, 051101. [Google Scholar] [CrossRef] [PubMed]
- Kealhofer, C.; Schneider, W.; Ehberger, D.; Ryabov, A.; Krausz, F.; Baum, P. All-Optical Control and Metrology of Electron Pulses. Science 2016, 352, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Wangler, T.P. RF Linear Accelerators, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Murooka, Y.; Naruse, N.; Sakakihara, S.; Ishimaru, M.; Yang, J.; Tanimura, K. Transmission-Electron Diffraction by MeV Electron Pulses. Appl. Phys. Lett. 2011, 98, 251903. [Google Scholar] [CrossRef]
- Hachmann, M.; Flöttmann, K. Measurement of Ultra Low Transverse Emittance at REGAE. Nucl. Instrum. Methods Phys. Res. A 2016, 829, 318–320. [Google Scholar] [CrossRef]
- Manz, S.; Casandruc, A.; Zhang, D.; Zhong, Y.; Loch, R.A.; Marx, A.; Hasegawa, T.; Liu, L.C.; Bayesteh, S.; Delsim-Hashemi, H.; et al. Mapping Atomic Motions with Ultrabright Electrons: Towards Fundamental Limits in Space-Time Resolution. Faraday Discuss. 2015, 177, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Weathersby, S.P.; Brown, G.; Centurion, M.; Chase, T.F.; Coffee, R.; Corbett, J.; Eichner, J.P.; Frisch, J.C.; Fry, A.R.; Gühr, M.; et al. Mega-Electron-Volt Ultrafast Electron Diffraction at SLAC National Accelerator Laboratory. Rev. Sci. Instrum. 2015, 86, 073702. [Google Scholar] [CrossRef]
- Williamson, C.J.; Zewail, A.H. Ultrafast Electron Diffraction. Velocity Mismatch and Temporal Resolution in Crossed-Beam Experiments. Chem. Phys. Lett. 1993, 209, 10–16. [Google Scholar] [CrossRef]
- Hommelhoff, P.; Sortais, Y.; Aghajani-Talesh, A.; Kasevich, M.A. Field Emission Tip as a Nanometer Source of Free Electron Femtosecond Pulses. Phys. Rev. Lett. 2006, 96, 077401. [Google Scholar] [CrossRef]
- Hommelhoff, P.; Kealhofer, C.; Kasevich, M.A. Ultrafast Electron Pulses from a Tungsten Tip Triggered by Low-Power Femtosecond Laser Pulses. Phys. Rev. Lett. 2006, 97, 247402. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.; Corder, C.; Strohaber, J.; Chandler-Smith, N.; Uiterwaal, C.; Batelaan, H. Laser-Induced Ultrafast Electron Emission from a Field Emission Tip. New J. Phys. 2007, 9, 2471. [Google Scholar] [CrossRef]
- Ropers, C.; Solli, D.R.; Schulz, C.P.; Lienau, C.; Elsaesser, T. Localized Multiphoton Emission of Femtosecond Electron Pulses from Metal Nanotips. Phys. Rev. Lett. 2007, 98, 043907. [Google Scholar] [CrossRef] [PubMed]
- Ropers, C.; Elsaesser, T.; Cerullo, G.; Zavelani-Rossi, M.; Lienau, C. Ultrafast Optical Excitations of Metallic Nanostructures: From Light Confinement to a Novel Electron Source. New J. Phys. 2007, 9, 397. [Google Scholar] [CrossRef]
- Paarmann, A.; Gulde, M.; Müller, M.; Schäfer, S.; Schweda, S.; Maiti, M.; Xu, C.; Hohage, T.; Schenk, F.; Ropers, C.; et al. Coherent Femtosecond Low-Energy Single-Electron Pulses for Time-Resolved Diffraction and Imaging: A Numerical Study. J. Appl. Phys. 2012, 112, 113109. [Google Scholar] [CrossRef]
- Hoffrogge, J.; Stein, J.P.; Krüger, M.; Förster, M.; Hammer, J.; Ehberger, D.; Baum, P.; Hommelhoff, P. Tip-Based Source of Femtosecond Electron Pulses at 30 KeV. J. Appl. Phys. 2014, 115, 094506. [Google Scholar] [CrossRef]
- Müller, M.; Paarmann, A.; Ernstorfer, R. Femtosecond Electrons Probing Currents and Atomic Structure in Nanomaterials. Nat. Commun. 2014, 5, 5292. [Google Scholar] [CrossRef]
- Gulde, M.; Schweda, S.; Storeck, G.; Maiti, M.; Yu, H.K.; Wodtke, A.M.; Schäfer, S.; Ropers, C. Ultrafast Low-Energy Electron Diffraction in Transmission Resolves Polymer/Graphene Superstructure Dynamics. Science 2014, 345, 200–204. [Google Scholar] [CrossRef]
- Storeck, G.; Vogelgesang, S.; Sivis, M.; Schäfer, S.; Ropers, C. Nanotip-Based Photoelectron Microgun for Ultrafast LEED. Struct. Dyn. 2017, 4, 044024. [Google Scholar] [CrossRef]
- Vogelgesang, S.; Storeck, G.; Horstmann, J.G.; Diekmann, T.; Sivis, M.; Schramm, S.; Rossnagel, K.; Schäfer, S.; Ropers, C. Phase Ordering of Charge Density Waves Traced by Ultrafast Low-Energy Electron Diffraction. Nat. Phys. 2018, 14, 184–190. [Google Scholar] [CrossRef]
- Scheinfein, M.R.; Qian, W.; Spence, J.C.H. Aberrations of Emission Cathodes: Nanometer Diameter Field-emission Electron Sources. J. Appl. Phys. 1993, 73, 2057–2068. [Google Scholar] [CrossRef]
- Qian, W.; Scheinfein, M.R.; Spence, J.C.H. Brightness Measurements of Nanometer-sized Field-emission-electron Sources. J. Appl. Phys. 1993, 73, 7041–7045. [Google Scholar] [CrossRef]
- Chang, C.-C.; Kuo, H.-S.; Hwang, I.-S.; Tsong, T.T. A Fully Coherent Electron Beam from a Noble-Metal Covered W(111) Single-Atom Emitter. Nanotechnology 2009, 20, 115401. [Google Scholar] [CrossRef] [PubMed]
- Steinwand, E.; Longchamp, J.-N.; Fink, H.-W. Coherent Low-Energy Electron Diffraction on Individual Nanometer Sized Objects. Ultramicroscopy 2011, 111, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Katsouleas, T.; Dawson, J.M. Unlimited Electron Acceleration in Laser-Driven Plasma Waves. Phys. Rev. Lett. 1983, 51, 392–395. [Google Scholar] [CrossRef]
- Faure, J.; Glinec, Y.; Pukhov, A.; Kiselev, S.; Gordienko, S.; Lefebvre, E.; Rousseau, J.-P.; Burgy, F.; Malka, V. A Laser–Plasma Accelerator Producing Monoenergetic Electron Beams. Nature 2004, 431, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.; Antici, P.; d’Humières, E.; Lefebvre, E.; Borghesi, M.; Brambrink, E.; Cecchetti, C.A.; Kaluza, M.; Malka, V.; Manclossi, M.; et al. Laser-Driven Proton Scaling Laws and New Paths towards Energy Increase. Nat. Phys. 2006, 2, 48–54. [Google Scholar] [CrossRef]
- Esarey, E.; Schroeder, C.B.; Leemans, W.P. Physics of Laser-Driven Plasma-Based Electron Accelerators. Rev. Mod. Phys. 2009, 81, 1229–1285. [Google Scholar] [CrossRef]
- Buck, A.; Nicolai, M.; Schmid, K.; Sears, C.M.S.; Sävert, A.; Mikhailova, J.M.; Krausz, F.; Kaluza, M.C.; Veisz, L. Real-Time Observation of Laser-Driven Electron Acceleration. Nat. Phys. 2011, 7, 543–548. [Google Scholar] [CrossRef]
- Lundh, O.; Lim, J.; Rechatin, C.; Ammoura, L.; Ben-Ismaïl, A.; Davoine, X.; Gallot, G.; Goddet, J.-P.; Lefebvre, E.; Malka, V.; et al. Few Femtosecond, Few Kiloampere Electron Bunch Produced by a Laser-Plasma Accelerator. Nat. Phys. 2011, 7, 219–222. [Google Scholar] [CrossRef]
- Marceau, V.; Varin, C.; Brabec, T.; Piché, M. Femtosecond 240-KeV Electron Pulses from Direct Laser Acceleration in a Low-Density Gas. Phys. Rev. Lett. 2013, 111, 224801. [Google Scholar] [CrossRef] [PubMed]
- Taban, G.; Reijnders, M.P.; Bell, S.C.; van der Geer, S.B.; Luiten, O.J.; Vredenbregt, E.J.D. Design and Validation of an Accelerator for an Ultracold Electron Source. Phys. Rev. Spec. Top. Accel. Beams 2008, 11, 050102. [Google Scholar] [CrossRef]
- McCulloch, A.J.; Sheludko, D.V.; Saliba, S.D.; Bell, S.C.; Junker, M.; Nugent, K.A.; Scholten, R.E. Arbitrarily Shaped High-Coherence Electron Bunches from Cold Atoms. Nat. Phys. 2011, 7, 785–788. [Google Scholar] [CrossRef]
- Saliba, S.D.; Putkunz, C.T.; Sheludko, D.V.; McCulloch, A.J.; Nugent, K.A.; Scholten, R.E. Spatial Coherence of Electron Bunches Extracted from an Arbitrarily Shaped Cold Atom Electron Source. Opt. Express 2012, 20, 3967. [Google Scholar] [CrossRef] [PubMed]
- Engelen, W.J.; van der Heijden, M.A.; Bakker, D.J.; Vredenbregt, E.J.D.; Luiten, O.J. High-Coherence Electron Bunches Produced by Femtosecond Photoionization. Nat. Commun. 2013, 4, 1693. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, A.J.; Sheludko, D.V.; Junker, M.; Scholten, R.E. High-Coherence Picosecond Electron Bunches from Cold Atoms. Nat. Commun. 2013, 4, 1692. [Google Scholar] [CrossRef] [PubMed]
- Van Mourik, M.W.; Engelen, W.J.; Vredenbregt, E.J.D.; Luiten, O.J. Ultrafast Electron Diffraction Using an Ultracold Source. Struct. Dyn. 2014, 1, 034302. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.J.; Murphy, D.; Speirs, R.W.; van Bijnen, R.M.W.; McCulloch, A.J.; Scholten, R.E.; Sparkes, B.M. Suppression of Emittance Growth Using a Shaped Cold Atom Electron and Ion Source. Phys. Rev. Lett. 2016, 117, 193202. [Google Scholar] [CrossRef]
- Torrance, J.S.; Speirs, R.W.; McCulloch, A.J.; Scholten, R.E. Time-Resolved Brightness Measurements by Streaking. Phys. Rev. Spec. Top. Accel. Beams 2018, 21, 032802. [Google Scholar] [CrossRef]
- Bostanjoglo, O.; Tornow, R.P.; Tornow, W. Nanosecond Transmission Electron Microscopy and Diffraction. J. Phys. E Sci. Instrum. 1987, 20, 556. [Google Scholar] [CrossRef]
- Kleinschmidt, H.; Ziegler, A.; Campbell, G.H.; Colvin, J.D.; Bostanjoglo, O. Phase Transformation Analysis in Titanium at Nanosecond Time Resolution. J. Appl. Phys. 2005, 98, 054313. [Google Scholar] [CrossRef]
- Armstrong, M.R.; Boyden, K.; Browning, N.D.; Campbell, G.H.; Colvin, J.D.; DeHope, W.J.; Frank, A.M.; Gibson, D.J.; Hartemann, F.; Kim, J.S.; et al. Practical Considerations for High Spatial and Temporal Resolution Dynamic Transmission Electron Microscopy. Ultramicroscopy 2007, 107, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; LaGrange, T.; Reed, B.W.; Taheri, M.L.; Armstrong, M.R.; King, W.E.; Browning, N.D.; Campbell, G.H. Imaging of Transient Structures Using Nanosecond in Situ TEM. Science 2008, 321, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.-H.; Zewail, A.H. 4D Electron Tomography. Science 2010, 328, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.; Flannigan, D.J.; Zewail, A.H. Photon-Induced Near-Field Electron Microscopy. Nature 2009, 462, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Kwon, O.-H.; Zewail, A.H. Dynamics of Chemical Bonding Mapped by Energy-Resolved 4D Electron Microscopy. Science 2009, 325, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, D.J.; Zewail, A.H. 4D Electron Microscopy: Principles and Applications. Acc. Chem. Res. 2012, 45, 1828–1839. [Google Scholar] [CrossRef]























© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciaini, G. Recent Advances in Ultrafast Structural Techniques. Appl. Sci. 2019, 9, 1427. https://doi.org/10.3390/app9071427
Sciaini G. Recent Advances in Ultrafast Structural Techniques. Applied Sciences. 2019; 9(7):1427. https://doi.org/10.3390/app9071427
Chicago/Turabian StyleSciaini, Germán. 2019. "Recent Advances in Ultrafast Structural Techniques" Applied Sciences 9, no. 7: 1427. https://doi.org/10.3390/app9071427
APA StyleSciaini, G. (2019). Recent Advances in Ultrafast Structural Techniques. Applied Sciences, 9(7), 1427. https://doi.org/10.3390/app9071427