Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer


Abstract
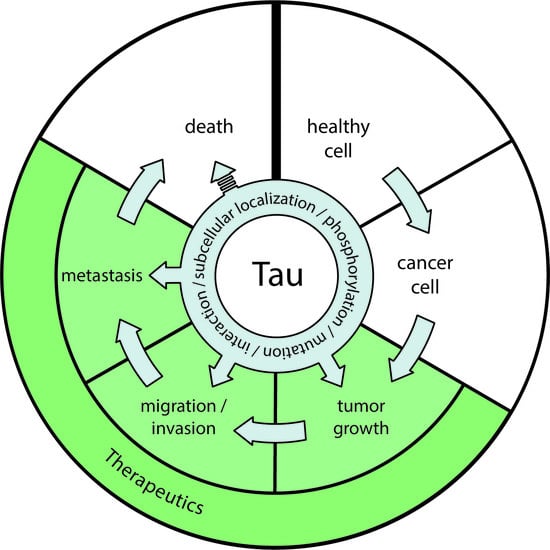
:
{kind=link}
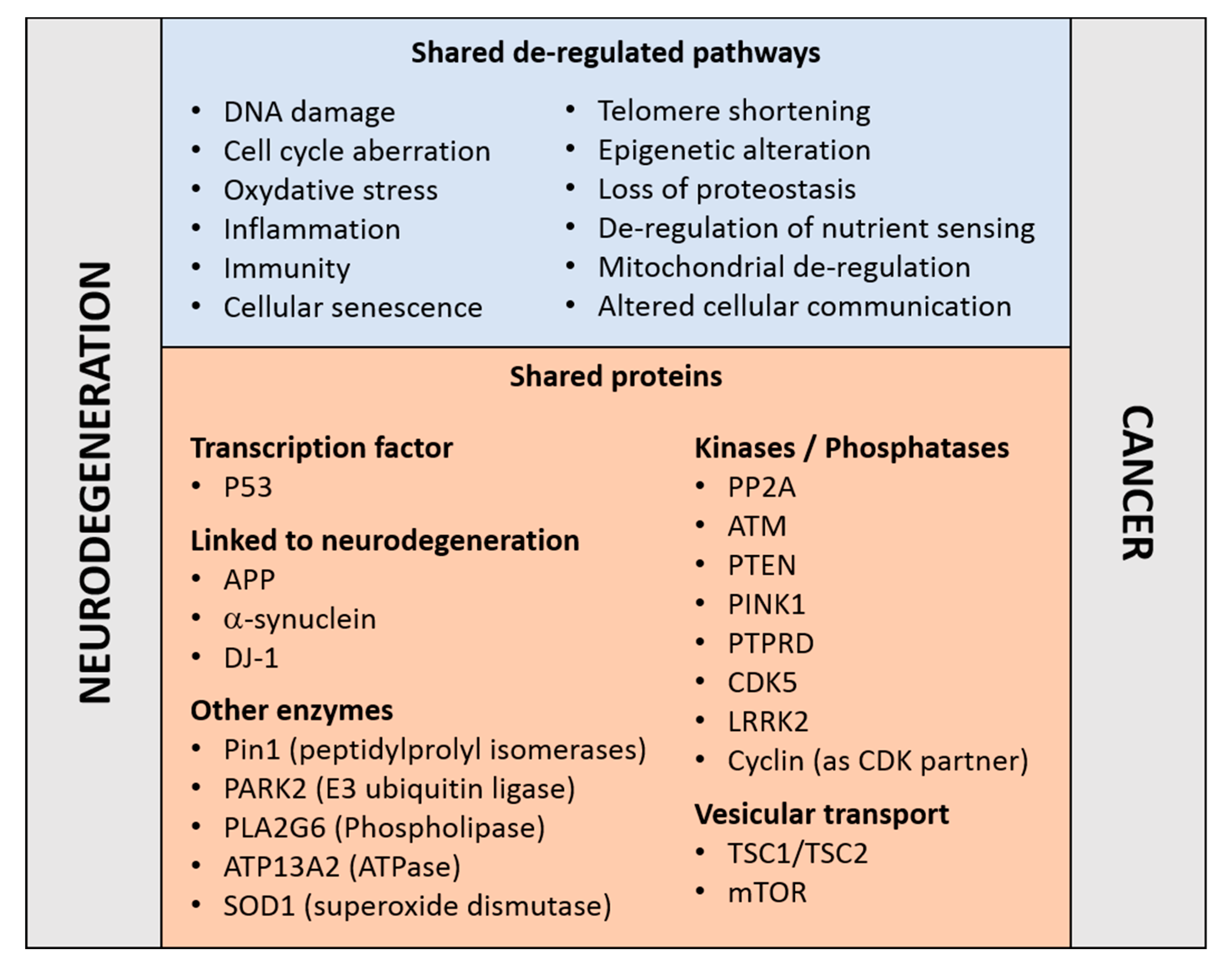{kind=link}
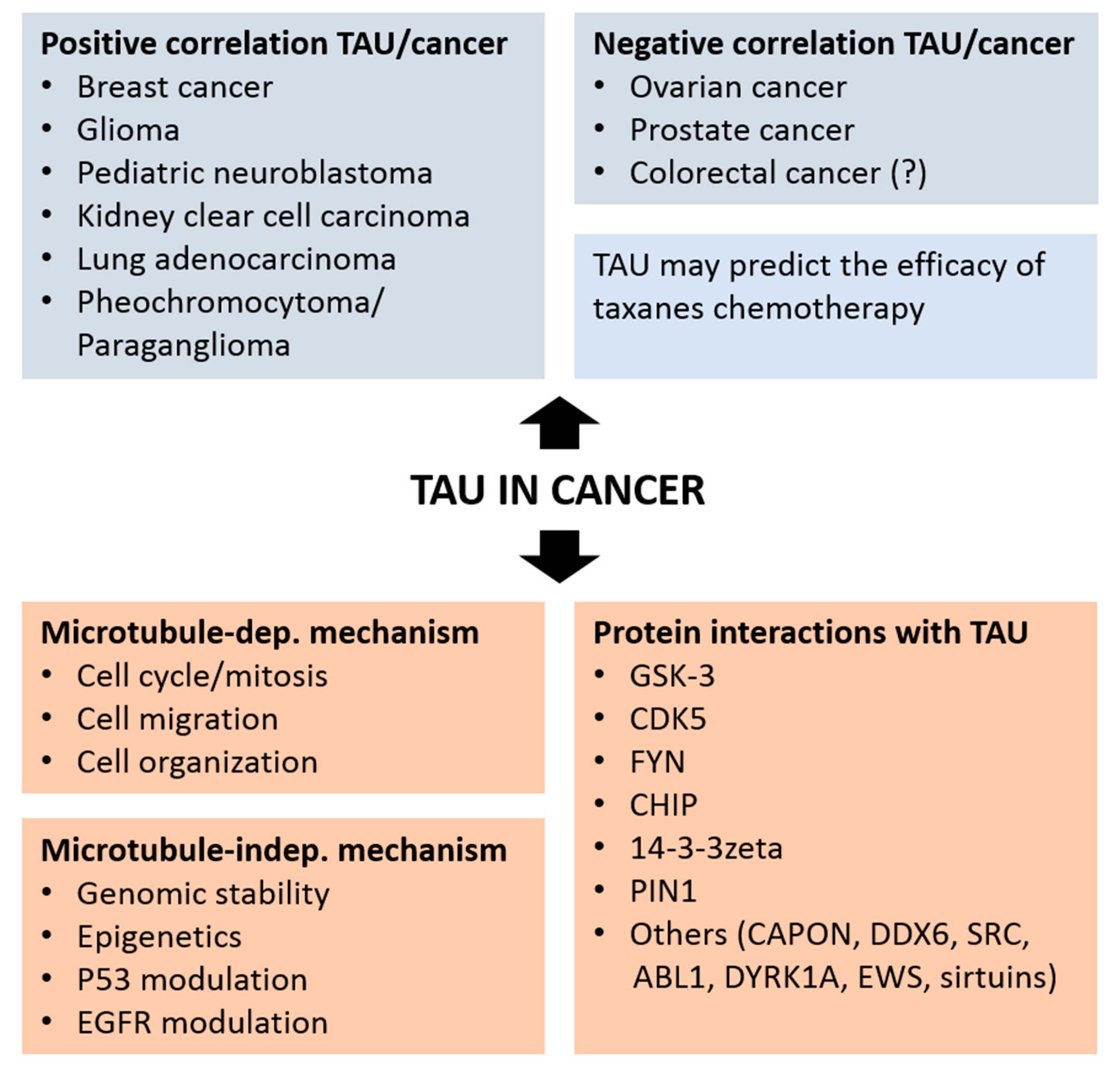{kind=link}
1. Coming Together: Cancer and Neurodegenerative Disorders, Do They Share Dysregulated Pathways?
2. The TAU Protein
3. TAU and Microtubule-Targeting Chemotherapy
4. TAU as a Prognostic Marker in Cancer
5. Possible Microtubules-Associated Mechanisms Explaining the Link between TAU and Cancer
6. Possible Microtubules-Independent Pathways Explaining the Link between TAU and Cancer
7. Protein-Protein Interactions Linking TAU to Cancer
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| ATM | ataxia-telangiectasia mutated |
| BioGRID | biological general repository for interaction datasets |
| CDK5 | cyclin-dependent kinase 5 |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| ER | estrogen receptor |
| ERG | erythroblast transformation specific-related gene |
| GBM | glioblastoma |
| GSK-3 | glycogen synthase kinase-3 |
| HD | Huntington’s disease |
| HDAC | histone deacetylase |
| HER2 | human epidermal growth factor receptor 2 |
| HIF | hypoxia-inducible factor |
| HSP | heat shock protein |
| IDH | isocitrate dehydrogenase |
| NMDA | N-methyl-D-aspartate |
| PD | Parkinson’s disease |
| PI3K | phosphoinositide 3 kinase |
| PR | progesterone receptor |
| TCGA | the cancer genome atlas |
References
- Currais, A.; Goldberg, J.; Farrokhi, C.; Chang, M.; Prior, M.; Dargusch, R.; Daugherty, D.; Armando, A.; Quehenberger, O.; Maher, P.; et al. A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging 2015, 7, 937–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Lee, H.P.; Kudo, W.; Zhu, X.; Smith, M.A.; Lee, H.G. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev. Mol. Med. 2010, 12, e19. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Lee, H.G.; Petersen, R.B.; Casadesus, G.; Vincent, I.; Linford, N.J.; Funk, J.O.; Shapiro, R.A.; Smith, M.A. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta 2007, 1772, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullaart, E.; Boerrigter, M.E.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol. Aging 1990, 11, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Bretaud, S.; Allen, C.; Ingham, P.W.; Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 2007, 100, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Meimaridou, E.; Tavassoli, M.; Melino, G.; Lovestone, S.; Killick, R. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci. Lett. 2007, 418, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1850, 2069–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef] [Green Version]
- Driver, J.A.; Logroscino, G.; Buring, J.E.; Gaziano, J.M.; Kurth, T. A prospective cohort study of cancer incidence following the diagnosis of Parkinson’s disease. Cancer Epidemiol. Biomark. 2007, 16, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Inzelberg, R.; Jankovic, J. Are Parkinson disease patients protected from some but not all cancers? Neurology 2007, 69, 1542–1550. [Google Scholar] [CrossRef]
- Møller, H.; Mellemkjaer, L.; McLaughlin, J.K.; Olsen, J.H. Occurrence of different cancers in patients with Parkinson’s disease. BMJ 1995, 310, 1500–1501. [Google Scholar] [CrossRef] [Green Version]
- Roe, C.M.; Fitzpatrick, A.L.; Xiong, C.; Sieh, W.; Kuller, L.; Miller, J.P.; Williams, M.M.; Kopan, R.; Behrens, M.I.; Morris, J.C. Cancer linked to Alzheimer disease but not vascular dementia. Neurology 2010, 74, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.B.; Tang, B.; Liu, Y.W.; Wang, X.F.; Chen, G.J. Alzheimer disease and cancer risk: A meta-analysis. J. Cancer Res. Clin. Oncol. 2015, 141, 485–494. [Google Scholar] [CrossRef]
- Sørensen, S.A.; Fenger, K.; Olsen, J.H. Significantly lower incidence of cancer among patients with Huntington disease: An apoptotic effect of an expanded polyglutamine tract? Cancer 1999, 86, 1342–1346. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martínez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Majd, S.; Power, J.; Majd, Z. Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, A.; Sanchez Valle, J.; Pancaldi, V.; Baudot, A.; Barillot, E.; Caselle, M.; Valencia, A.; Zinovyev, A.; Cantini, L. Molecular Inverse Comorbidity between Alzheimer’s Disease and Lung Cancer: New Insights from Matrix Factorization. Int. J. Mol. Sci. 2019, 20, 3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology 2013, 81, 322–328. [Google Scholar] [CrossRef]
- Ou, S.M.; Lee, Y.J.; Hu, Y.W.; Liu, C.J.; Chen, T.J.; Fuh, J.L.; Wang, S.J. Does Alzheimer’s Disease Protect against Cancers? A Nationwide Population-Based Study. Neuroepidemiology 2013, 40, 42–49. [Google Scholar] [CrossRef]
- Sánchez-Valle, J.; Tejero, H.; Ibáñez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474. [Google Scholar] [CrossRef] [Green Version]
- Gibson, S.B.; Abbott, D.; Farnham, J.M.; Thai, K.K.; McLean, H.; Figueroa, K.P.; Bromberg, M.B.; Pulst, S.M.; Cannon-Albright, L. Population-based risks for cancer in patients with ALS. Neurology 2016, 87, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Al-Chalabi, A.; Ronnevi, L.O.; Turner, M.R.; Wirdefeldt, K.; Kamel, F.; Ye, W. Amyotrophic lateral sclerosis and cancer: A register-based study in Sweden. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, D.M.; Wu, J.; Daugherty, S.E.; Kuncl, R.W.; Enewold, L.R.; Pfeiffer, R.M. The risk of amyotrophic lateral sclerosis after cancer in U.S. elderly adults: A population-based prospective study. Int. J. Cancer 2014, 135, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Li, X.; Jankovic, J. The association between Parkinson’s disease and melanoma. Int. J. Cancer 2011, 128, 2251–2260. [Google Scholar] [CrossRef]
- Hu, H.-H.; Kannengiesser, C.; Lesage, S.; André, J.; Mourah, S.; Michel, L.; Descamps, V.; Basset-Seguin, N.; Bagot, M.; Bensussan, A.; et al. PARKIN Inactivation Links Parkinson’s Disease to Melanoma. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef] [Green Version]
- Kareus, S.A.; Figueroa, K.P.; Cannon-Albright, L.A.; Pulst, S.M. Shared predispositions of parkinsonism and cancer: A population-based pedigree-linked study. Arch. Neurol. 2012, 69, 1572–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Gao, X.; Lu, Y.; Chen, H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 2011, 76, 2002–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, U.; Heilmann, E.; Voss, J.; Riedel, K.; Zhivov, A.; Schäd, S.G.; Gross, G.E.; Benecke, R.; Trcka, J. Frequency and profile of Parkinson’s disease prodromi in patients with malignant melanoma. J. Neurol. Neurosurg. Psychiatry 2016, 87, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Frain, L.; Swanson, D.; Cho, K.; Gagnon, D.; Lu, K.P.; Betensky, R.A.; Driver, J. Association of cancer and Alzheimer’s disease risk in a national cohort of veterans. Alzheimer’s Dement. 2017, 13, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Kesler, S.R.; Watson, C.L.; Blayney, D.W. Brain network alterations and vulnerability to simulated neurodegeneration in breast cancer. Neurobiol. Aging 2015, 36, 2429–2442. [Google Scholar] [CrossRef] [Green Version]
- Ganguli, M. Cancer and Dementia: It’s Complicated. Alzheimer Dis. Assoc. Disord. 2015, 29, 177–182. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef]
- Benilova, I.; De Strooper, B. Prion protein in Alzheimer’s pathogenesis: A hot and controversial issue. EMBO Mol. Med. 2010, 2, 289–290. [Google Scholar] [CrossRef]
- Driver, J.A. Inverse association between cancer and neurodegenerative disease: Review of the epidemiologic and biological evidence. Biogerontology 2014, 15, 547–557. [Google Scholar] [CrossRef]
- Du, L.; Pertsemlidis, A. Cancer and neurodegenerative disorders: Pathogenic convergence through microRNA regulation. J. Mol. Cell Biol. 2011, 3, 176–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klus, P.; Cirillo, D.; Botta Orfila, T.; Gaetano Tartaglia, G. Neurodegeneration and Cancer: Where the Disorder Prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plun-Favreau, H.; Lewis, P.A.; Hardy, J.; Martins, L.M.; Wood, N.W. Cancer and neurodegeneration: Between the devil and the deep blue sea. PLoS Genet. 2010, 6, e1001257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, L.G.T.; Veeriah, S.; Chan, T.A. Genetic determinants at the interface of cancer and neurodegenerative disease. Oncogene 2010, 29, 3453–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; Kraft, P.; Cormack, J.; Blalock, K.; Campbell, P.T.; Casey, G.; Conti, D.V.; Edlund, C.K.; et al. Investigating the genetic relationship between Alzheimer’s disease and cancer using GWAS summary statistics. Hum. Genet. 2017, 136, 1341–1351. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, K.; Boullosa, C.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, S.; Lu, C. Amyloid precursor protein promotes the migration and invasion of breast cancer cells by regulating the MAPK signaling pathway. Int. J. Mol. Med. 2020, 45, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.Y.S.; Lee, M.A.; Ni, Y.-B.; Chan, S.-K.; Cheung, S.-Y.; Chan, W.-W.; Lau, K.-F.; Tse, G.M.K. Amyloid Precursor Protein Is Associated with Aggressive Behavior in Nonluminal Breast Cancers. Oncologist 2018, 23, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Xu, K. Alpha-synuclein contributes to malignant progression of human meningioma via the Akt/mTOR pathway. Cancer Cell Int. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Redaelli, V.; Contiero, P.; Fabiano, S.; Tagliabue, G.; Perego, P.; Benussi, L.; Bruni, A.C.; Filippini, G.; Farinotti, M.; et al. Tau Mutations Serve as a Novel Risk Factor for Cancer. Cancer Res. 2018, 78, 3731–3739. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Conconi, D.; Panzeri, E.; Redaelli, S.; Piccoli, E.; Paoletta, L.; Dalprà, L.; Tagliavini, F. Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J. Alzheimers Dis. 2013, 33, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 2005, 1739, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Tolnay, M.; Probst, A. REVIEW: Tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol. Appl. Neurobiol. 1999, 25, 171–187. [Google Scholar] [CrossRef]
- Yoshida, M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 2006, 26, 457–470. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. Inclusion-body myositis: Muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer’s and Parkinson’s disease brains. Acta Neuropathol. 2008, 116, 583–595. [Google Scholar] [CrossRef] [Green Version]
- François, M.; Leifert, W.; Martins, R.; Thomas, P.; Fenech, M. Biomarkers of Alzheimer’s disease risk in peripheral tissues; focus on buccal cells. Curr. Alzheimer Res. 2014, 11, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Hattori, H.; Matsumoto, M.; Iwai, K.; Tsuchiya, H.; Miyauchi, E.; Takasaki, M.; Kamino, K.; Munehira, J.; Kimura, Y.; Kawanishi, K.; et al. The tau protein of oral epithelium increases in Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2002, 57, M64–M70. [Google Scholar] [CrossRef] [Green Version]
- Ingelson, M.; Vanmechelen, E.; Lannfelt, L. Microtubule-associated protein tau in human fibroblasts with the Swedish Alzheimer mutation. Neurosci. Lett. 1996, 220, 9–12. [Google Scholar] [CrossRef]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Sui, Y.-T.; Peskind, E.R.; Li, G.; Hwang, H.; Devic, I.; Ginghina, C.; Edgar, J.S.; Pan, C.; Goodlett, D.R.; et al. Salivary tau species are potential biomarkers of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 299–305. [Google Scholar] [CrossRef]
- Souter, S.; Lee, G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J. Cell. Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Dugger, B.N.; Whiteside, C.M.; Maarouf, C.L.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Garcia, A.; Dunckley, T.; Meechoovet, B.; Reiman, E.M.; et al. The Presence of Select Tau Species in Human Peripheral Tissues and Their Relation to Alzheimer’s Disease. J. Alzheimers Dis. 2016, 51, 345–356. [Google Scholar] [CrossRef]
- Gu, Y.; Oyama, F.; Ihara, Y. τ Is Widely Expressed in Rat Tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef]
- Boyne, L.J.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef]
- Couchie, D.; Mavilia, C.; Georgieff, I.S.; Liem, R.K.; Shelanski, M.L.; Nunez, J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 4378–4381. [Google Scholar] [CrossRef] [Green Version]
- Lau, D.H.W.; Hogseth, M.; Phillips, E.C.; O’Neill, M.J.; Pooler, A.M.; Noble, W.; Hanger, D.P. Critical residues involved in tau binding to fyn: Implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Loomis, P.A.; Howard, T.H.; Castleberry, R.P.; Binder, L.I. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Loomis, P.A.; Zinkowski, R.P.; Binder, L.I. A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J. Cell Biol. 1993, 121, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a Key Player in Neuronal DNA Protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [Green Version]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2019, 101, 349. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Sola, M.; Magrin, C.; Pedrioli, G.; Pinton, S.; Salvadè, A.; Papin, S.; Paganetti, P. Tau affects P53 function and cell fate during the DNA damage response. Commun. Biol. 2020, 3, 245. [Google Scholar] [CrossRef]
- Ferreira, C.G.; Tolis, C.; Giaccone, G. p53 and chemosensitivity. Ann. Oncol. 1999, 10, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Taxanes for the treatment of metastatic breast cancer. Breast Cancer 2012, 6, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, L.M.; Sampath, D. Resistance To Taxanes. In Cancer Drug Resistance; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 329–358. [Google Scholar] [CrossRef]
- Kar, S.; Fan, J.; Smith, M.J.; Goedert, M.; Amos, L.A. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Taira, N.; Hara, F.; Fujita, T.; Yamamoto, H.; Soh, J.; Toyooka, S.; Nogami, T.; Shien, T.; Doihara, H.; et al. The estrogen receptor influences microtubule-associated protein tau (MAPT) expression and the selective estrogen receptor inhibitor fulvestrant downregulates MAPT and increases the sensitivity to taxane in breast cancer cells. Breast Cancer Res. 2010, 12, R43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoter, M.; Bodnar, L.; Duchnowska, R.; Stec, R.; Grala, B.; Szczylik, C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother. Pharmacol. 2011, 68, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, P.; Wang, B.; Clark, E.; Lee, H.; Rouzier, R.; Pusztai, L. Microtubule Associated Protein (MAP)-Tau: A Novel Mediator of Paclitaxel Sensitivity In Vitro and In Vivo. Cell Cycle 2005, 4, 1149–1152. [Google Scholar] [CrossRef]
- Smith, M.J.; Crowther, R.A.; Goedert, M. The natural osmolyte trimethylamine N-oxide (TMAO) restores the ability of mutant tau to promote microtubule assembly. FEBS Lett. 2000, 484, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Valet, F.; de Cremoux, P.; Spyratos, F.; Servant, N.; Dujaric, M.E.; Gentien, D.; Lehmann-Che, J.; Scott, V.; Sigal-Zafrani, B.; Mathieu, M.C.; et al. Challenging single- and multi-probesets gene expression signatures of pathological complete response to neoadjuvant chemotherapy in breast cancer: Experience of the REMAGUS 02 phase II trial. Breast 2013, 22, 1052–1059. [Google Scholar] [CrossRef]
- Smoter, M.; Bodnar, L.; Grala, B.; Stec, R.; Zieniuk, K.; Kozlowski, W.; Szczylik, C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitaxel/platinum first-line chemotherapy. J. Exp. Clin. Cancer Res. 2013, 32, 25. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, A.; Kobayashi, A.; Oikiri, H.; Yokoyama, Y. Functional role of the Tau protein in epithelial ovarian cancer cells. Reprod. Med. Biol. 2017, 16, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Mimori, K.; Sadanaga, N.; Yoshikawa, Y.; Ishikawa, K.; Hashimoto, M.; Tanaka, F.; Sasaki, A.; Inoue, H.; Sugimachi, K.; Mori, M. Reduced tau expression in gastric cancer can identify candidates for successful Paclitaxel treatment. Br. J. Cancer 2006, 94, 1894–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangrajrang, S.; Denoulet, P.; Millot, G.; Tatoud, R.; Podgorniak, M.P.; Tew, K.D.; Calvo, F.; Fellous, A. Estramustine resistance correlates with tau over-expression in human prostatic carcinoma cells. Int. J. Cancer 1998, 77, 626–631. [Google Scholar] [CrossRef]
- Yoo, J.; Shim, B.Y.; Yoo, C.Y.; Kang, S.J.; Lee, K.Y. Predictive Significance of KRAS and Tau for Chemoresponse in Advanced Non-Small-Cell Lung Cancer. J. Pathol. Transl. Med. 2009, 43, 435–440. [Google Scholar] [CrossRef]
- Guise, S.; Braguer, D.; Remacle-Bonnet, M.; Pommier, G.; Briand, C. Tau protein is involved in the apoptotic process induced by anti-microtubule agents on neuroblastoma cells. Apoptosis 1999, 4, 47–58. [Google Scholar] [CrossRef]
- Baquero, M.T.; Lostritto, K.; Gustavson, M.D.; Bassi, K.A.; Appia, F.; Camp, R.L.; Molinaro, A.M.; Harris, L.N.; Rimm, D.L. Evaluation of prognostic and predictive value of microtubule associated protein tau in two independent cohorts. Breast Cancer Res. 2011, 13, R85. [Google Scholar] [CrossRef] [Green Version]
- Pentheroudakis, G.; Kalogeras, K.T.; Wirtz, R.M.; Grimani, I.; Zografos, G.; Gogas, H.; Stropp, U.; Pectasides, D.; Skarlos, D.; Hennig, G.; et al. Gene expression of estrogen receptor, progesterone receptor and microtubule-associated protein Tau in high-risk early breast cancer: A quest for molecular predictors of treatment benefit in the context of a Hellenic Cooperative Oncology Group trial. Breast Cancer Res. Treat. 2009, 116, 131–143. [Google Scholar] [CrossRef]
- Pusztai, L.; Jeong, J.-H.; Gong, Y.; Ross, J.S.; Kim, C.; Paik, S.; Rouzier, R.; Andre, F.; Hortobagyi, G.N.; Wolmark, N.; et al. Evaluation of microtubule-associated protein-Tau expression as a prognostic and predictive marker in the NSABP-B 28 randomized clinical trial. J. Clin. Oncol. 2009, 27, 4287–4292. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Y.; Liu, W.; Li, Z.; Wei, Z.; Jiang, R. Microtubule-associated protein tau is associated with the resistance to docetaxel in prostate cancer cell lines. Res. Rep. Urol. 2017, 9, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Huang, M.; Lu, M.; Zhu, W.; Shu, Y.; Cao, P.; Liu, P. Regulation of microtubule-associated protein tau (MAPT) by miR-34c-5p determines the chemosensitivity of gastric cancer to paclitaxel. Cancer Chemother. Pharmacol. 2013, 71, 1159–1171. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Z.; Sun, L.; Fang, Y.; Xu, X.; Zhou, G. miR-186 regulates chemo-sensitivity to paclitaxel via targeting MAPT in non-small cell lung cancer (NSCLC). Mol. Biosyst. 2016, 12, 3417–3424. [Google Scholar] [CrossRef]
- Hristodorov, D.; Mladenov, R.; Pardo, A.; Pham, A.T.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br. J. Cancer 2013, 109, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinrinmade, O.A.; Jordaan, S.; Hristodorov, D.; Mladenov, R.; Mungra, N.; Chetty, S.; Barth, S. Human MAP Tau Based Targeted Cytolytic Fusion Proteins. Biomedicines 2017, 5, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneau, C.; Gurard-Levin, Z.A.; Andre, F.; Pusztai, L.; Rouzier, R. Predictive and Prognostic Value of the TauProtein in Breast Cancer. Anticancer Res. 2015, 35, 5179–5184. [Google Scholar] [PubMed]
- Shao, Y.Y.; Kuo, K.T.; Hu, F.C.; Lu, Y.S.; Huang, C.S.; Liau, J.Y.; Lee, W.C.; Hsu, C.; Kuo, W.H.; Chang, K.J.; et al. Predictive and prognostic values of tau and ERCC1 in advanced breast cancer patients treated with paclitaxel and cisplatin. Jpn. J. Clin. Oncol. 2010, 40, 286–293. [Google Scholar] [CrossRef]
- Andre, F.; Hatzis, C.; Anderson, K.; Sotiriou, C.; Mazouni, C.; Mejia, J.; Wang, B.; Hortobagyi, G.N.; Symmans, W.F.; Pusztai, L. Microtubule-associated protein-tau is a bifunctional predictor of endocrine sensitivity and chemotherapy resistance in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2007, 13, 2061–2067. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Caceres, A. Estrogen-enhanced neurite growth: Evidence for a selective induction of Tau and stable microtubules. J. Neurosci. 1991, 11, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004, 64, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Lew, G.M. Changes in microtubular tau protein after estrogen in a cultured human neuroblastoma cell line. Gen. Pharmacol. 1993, 24, 1383–1386. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Spreafico, E.; Pollio, G.; Santagati, S.; Conti, E.; Cattaneo, E.; Maggi, A. Activated estrogen receptor mediates growth arrest and differentiation of a neuroblastoma cell line. Proc. Natl. Acad. Sci. USA 1993, 90, 3740–3744. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, A.; Takekoshi, S.; Sanno, N.; Utsunomiya, H.; Ohsugi, Y.; Saito, N.; Kanemitsu, H.; Tamura, A.; Nagashima, T.; Osamura, R.Y.; et al. Modulation of protein kinases and microtubule-associated proteins and changes in ultrastructure in female rat pituitary cells: Effects of estrogen and bromocriptine. J. Histochem. Cytochem. 1997, 45, 805–813. [Google Scholar] [CrossRef] [Green Version]
- West, M.; Blanchette, C.; Dressman, H.; Huang, E.; Ishida, S.; Spang, R.; Zuzan, H.; Olson, J.A.; Marks, J.R.; Nevins, J.R. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 11462–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Sui, M.; Huang, Y.; Park, B.H.; Davidson, N.E.; Fan, W. Estrogen receptor alpha mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res. 2007, 67, 5337–5344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Novel Functions of the Neurodegenerative-Related Gene Tau in Cancer. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matrone, M.A.; Whipple, R.A.; Thompson, K.; Cho, E.H.; Vitolo, M.I.; Balzer, E.M.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Tan, M.; et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene 2010, 29, 3217–3227. [Google Scholar] [CrossRef] [Green Version]
- Cirak, Y.; Sarsik, B.; Cakar, B.; Sen, S.; Simsir, A.; Uslu, R. Predictive and prognostic values of Tau and BubR1 protein in prostate cancer and their relationship to the Gleason score. Med Oncol. 2013, 30, 526. [Google Scholar] [CrossRef]
- Schroeder, C.; Grell, J.; Hube-Magg, C.; Kluth, M.; Lang, D.; Simon, R.; Höflmayer, D.; Minner, S.; Burandt, E.; Clauditz, T.S.; et al. Aberrant expression of the microtubule-associated protein tau is an independent prognostic feature in prostate cancer. BMC Cancer 2019, 19, 193. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Birdsey, G.M.; Dryden, N.H.; Shah, A.V.; Hannah, R.; Hall, M.D.; Haskard, D.O.; Parsons, M.; Mason, J.C.; Zvelebil, M.; Gottgens, B.; et al. The transcription factor Erg regulates expression of histone deacetylase 6 and multiple pathways involved in endothelial cell migration and angiogenesis. Blood 2012, 119, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Chow, A.; Amemiya, Y.; Sugar, L.; Nam, R.; Seth, A. Whole-transcriptome analysis reveals established and novel associations with TMPRSS2:ERG fusion in prostate cancer. Anticancer Res. 2012, 32, 3629–3641. [Google Scholar] [PubMed]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekino, Y.; Han, X.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Shiota, M.; Takeshima, Y.; Yasui, W.; et al. Microtubule-associated protein tau (MAPT) promotes bicalutamide resistance and is associated with survival in prostate cancer. Urol. Oncol. 2020, 38, 795.e791–795.e798. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Anderton, B.H.; Davis, D.R.; Gallo, J.M. Tau isoform expression and phosphorylation state during differentiation of cultured neuronal cells. FEBS Lett. 1995, 375, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Iqbal, K.; Trenkner, E.; Liu, D.J.; Grundke-Iqbal, I. Abnormally phosphorylated tau in SY5Y human neuroblastoma cells. FEBS Lett. 1995, 360, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Chobrutskiy, B.I.; Blanck, G. MAPT (Tau) expression is a biomarker for an increased rate of survival in pediatric neuroblastoma. Cell Cycle 2018, 17, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Chobrutskiy, B.I.; Sikaria, D.; Blanck, G. MAPT (Tau) expression is a biomarker for an increased rate of survival for low‑grade glioma. Oncol. Rep. 2019, 41, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; Segura-Collar, B.; Herránz, B.; García-Escudero, V.; Romero-Bravo, A.; Núñez, F.J.; García-Pérez, D.; Gutiérrez-Guamán, J.; Ayuso-Sacido, A.; Seoane, J.; et al. The IDH-TAU-EGFR triad defines the neovascular landscape of diffuse gliomas. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Guo, W.; Zhu, X.; Ahuja, N.; Fu, T. MAPT promoter CpG island hypermethylation is associated with poor prognosis in patients with stage II colorectal cancer. Cancer Manag. Res. 2019, 11, 7337–7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, A.; Nagata, K.; Hatsuta, H.; Takuma, H.; Bundo, M.; Iwamoto, K.; Tamaoka, A.; Murayama, S.; Saido, T.; Tsuji, S. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum. Mol. Genet. 2014, 23, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Coupland, K.G.; Mellick, G.D.; Silburn, P.A.; Mather, K.; Armstrong, N.J.; Sachdev, P.S.; Brodaty, H.; Huang, Y.; Halliday, G.M.; Hallupp, M.; et al. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro. Mov. Disord. 2014, 29, 1606–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shui, I.M.; Wong, C.J.; Zhao, S.; Kolb, S.; Ebot, E.M.; Geybels, M.S.; Rubicz, R.; Wright, J.L.; Lin, D.W.; Klotzle, B.; et al. Prostate tumor DNA methylation is associated with cigarette smoking and adverse prostate cancer outcomes. Cancer 2016, 122, 2168–2177. [Google Scholar] [CrossRef]
- Kit, O.I.; Vodolazhsky, D.I.; Kutilin, D.S.; Enin, Y.S.; Gevorkyan, Y.A.; Zolotukhin, P.V.; Boumber, Y.; Kharin, L.V.; Panina, S.B. A Proteomics Analysis Reveals 9 Up-Regulated Proteins Associated with Altered Cell Signaling in Colon Cancer Patients. Protein J. 2017, 36, 513–522. [Google Scholar] [CrossRef]
- Huda, M.N.; Erdene-Ochir, E.; Pan, C.H. Assay for Phosphorylation and Microtubule Binding Along with Localization of Tau Protein in Colorectal Cancer Cells. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Bou Samra, E.; Buhagiar-Labarchède, G.; Machon, C.; Guitton, J.; Onclercq-Delic, R.; Green, M.R.; Alibert, O.; Gazin, C.; Veaute, X.; Amor-Guéret, M. A role for Tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat. Commun. 2017, 8, 693. [Google Scholar] [CrossRef]
- Cirillo, L.; Gotta, M.; Meraldi, P. The Elephant in the Room: The Role of Microtubules in Cancer. Adv. Exp. Med. Biol. 2017, 1002, 93–124. [Google Scholar] [CrossRef]
- Hernandez, P.; Tirnauer, J.S. Tumor suppressor interactions with microtubules: Keeping cell polarity and cell division on track. Dis. Models Mech. 2010, 3, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Bougé, A.-L.; Parmentier, M.-L. Tau excess impairs mitosis and kinesin-5 function, leading to aneuploidy and cell death. Dis. Models Mech. 2016, 9, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, D.; Fischer, A.H.; Nickerson, J.A. Nuclear structure in cancer cells. Nat. Rev. Cancer 2004, 4, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Breuzard, G.; Pagano, A.; Bastonero, S.; Malesinski, S.; Parat, F.; Barbier, P.; Peyrot, V.; Kovacic, H. Tau regulates the microtubule-dependent migration of glioblastoma cells via the Rho-ROCK signaling pathway. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.-P.; Wang, G.-X.; Xia, P.; Hou, T.-T.; Zhou, H.-L.; Wang, T.-J.; Yang, X.-Y. Effects of microtubule-associated protein tau expression on neural stem cell migration after spinal cord injury. Neural Regen. Res. 2016, 11, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of p53 in neurodegenerative diseases. Neuro-Degener. Dis. 2012, 9, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational altered p53 as an early marker of oxidative stress in Alzheimer’s disease. PLoS ONE 2012, 7, e29789. [Google Scholar] [CrossRef]
- Stanga, S.; Lanni, C.; Sinforiani, E.; Mazzini, G.; Racchi, M. Searching for predictive blood biomarkers: Misfolded p53 in mild cognitive impairment. Curr. Alzheimer Res. 2012, 9, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Wang, S.; Song, J.; Jia, J. Combination of p53(ser15) and p21/p21(thr145) in peripheral blood lymphocytes as potential Alzheimer’s disease biomarkers. Neurosci. Lett. 2012, 516, 226–231. [Google Scholar] [CrossRef]
- Dorszewska, J.; Oczkowska, A.; Suwalska, M.; Rozycka, A.; Florczak-Wyspianska, J.; Dezor, M.; Lianeri, M.; Jagodzinski, P.P.; Kowalczyk, M.J.; Prendecki, M.; et al. Mutations in the exon 7 of Trp53 gene and the level of p53 protein in double transgenic mouse model of Alzheimer’s disease. Folia Neuropathol. 2014, 52, 30–40. [Google Scholar] [CrossRef]
- Cancino, G.I.; Yiu, A.P.; Fatt, M.P.; Dugani, C.B.; Flores, E.R.; Frankland, P.W.; Josselyn, S.A.; Miller, F.D.; Kaplan, D.R. p63 Regulates adult neural precursor and newly born neuron survival to control hippocampal-dependent Behavior. J. Neurosci. 2013, 33, 12569–12585. [Google Scholar] [CrossRef]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the ‘longevity-assurance’ isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.; Varriano, S.; Ordonez, M.; Kuczaj, P.; Murphy, M.R.; Aruggoda, G.; Lundine, D.; Morozova, V.; Makki, A.E.; Alonso, A.d.C.; et al. Nuclear Tau, p53 and Pin1 Regulate PARN-Mediated Deadenylation and Gene Expression. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huda, M.N.; Kim, D.H.; Erdene-Ochir, E.; Kim, Y.S.; Pan, C.-H. Expression, phosphorylation, localization, and microtubule binding of tau in colorectal cell lines. Appl. Biol. Chem. 2016, 59, 807–812. [Google Scholar] [CrossRef]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau Aggregates in Human Tauopathies. Brain Sci. 2019, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Iijima-Ando, K.; Zhao, L.; Gatt, A.; Shenton, C.; Iijima, K. A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Hum. Mol. Genet. 2010, 19, 1930–1938. [Google Scholar] [CrossRef] [Green Version]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.; Santa-Maria, I.; Gomez de Barreda, E.; Zhu, X.; Cuadros, R.; Cabrero, J.R.; Sanchez-Madrid, F.; Dawson, H.N.; Vitek, M.P.; Perry, G.; et al. Tau—An inhibitor of deacetylase HDAC6 function. J. Neurochem. 2009, 109, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.S.; Hubbert, C.C.; Yao, T.P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010, 285, 11219–11226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M.; Hernández, F.; Avila, J. New Features about Tau Function and Dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2018, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Wang, S.L.; Zhu, L.; Wu, P.Y.; Dai, W.B.; Rakesh, K.P. Structure-activity relationship (SAR) studies of synthetic glycogen synthase kinase-3β inhibitors: A critical review. Eur. J. Med. Chem. 2019, 164, 448–470. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-Dependent Protein Kinase 5 Primes Microtubule-Associated Protein Tau Site-Specifically for Glycogen Synthase Kinase 3β. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Shi, J.; Yin, D.; El-Akkad, E.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett. 2006, 580, 6269–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of phosphorylation of tau by cyclin-dependent kinase 5 and glycogen synthase kinase-3 at substrate level. FEBS Lett. 2006, 580, 5925–5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, R.; Zhang, J.; Zhang, J.; Lin, Z. Cyclin-dependent kinase 5 acts as a promising biomarker in clear cell Renal Cell Carcinoma. BMC Cancer 2019, 19, 698. [Google Scholar] [CrossRef]
- Feldmann, G.; Mishra, A.; Hong, S.-M.; Bisht, S.; Strock, C.J.; Ball, D.W.; Goggins, M.; Maitra, A.; Nelkin, B.D. Inhibiting the Cyclin-Dependent Kinase CDK5 Blocks Pancreatic Cancer Formation and Progression through the Suppression of Ras-Ral Signaling. Cancer Res. 2010, 70, 4460–4469. [Google Scholar] [CrossRef] [Green Version]
- Demelash, A.; Rudrabhatla, P.; Pant, H.C.; Wang, X.; Amin, N.D.; McWhite, C.D.; Naizhen, X.; Linnoila, R.I. Achaete-scute homologue-1 (ASH1) stimulates migration of lung cancer cells through Cdk5/p35 pathway. Mol. Biol. Cell 2012, 23, 2856–2866. [Google Scholar] [CrossRef]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, S.M.; Liebl, J.; Ardelt, M.A.; Lehr, T.; De Toni, E.N.; Mayr, D.; Brandl, L.; Kirchner, T.; Zahler, S.; Gerbes, A.L.; et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma--A novel therapeutic approach. J. Hepatol. 2015, 63, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Herzog, J.; Ehrlich, S.M.; Pfitzer, L.; Liebl, J.; Fröhlich, T.; Arnold, G.J.; Mikulits, W.; Haider, C.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 stabilizes hypoxia-inducible factor-1α: A novel approach for inhibiting angiogenesis in hepatocellular carcinoma. Oncotarget 2016, 7, 27108–27121. [Google Scholar] [CrossRef] [PubMed]
- Strock, C.J.; Park, J.-I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-Dependent Kinase 5 Activity Controls Cell Motility and Metastatic Potential of Prostate Cancer Cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, D.; Ditzel, H.J. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol. Res. 2015, 100, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Ghosh, M.K. A CHIPotle in physiology and disease. Int. J. Biochem. Cell Biol. 2015, 58, 37–52. [Google Scholar] [CrossRef]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef]
- McDonough, H.; Patterson, C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Zang, J.; Dai, H.-J.; Li, F.; Guo, F. Ubiquitin ligase CHIP functions as an oncogene and activates the AKT signaling pathway in prostate cancer. Int. J. Oncol. 2018, 53, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; He, J.; Wang, Y.; Feng, Q.; Zhang, Y.; Guo, J.; Li, J.; Li, S.; Wang, Y.; Yan, G.; et al. CHIP/Stub1 regulates the Warburg effect by promoting degradation of PKM2 in ovarian carcinoma. Oncogene 2017, 36, 4191–4200. [Google Scholar] [CrossRef]
- Yonezawa, T.; Takahashi, H.; Shikata, S.; Liu, X.; Tamura, M.; Asada, S.; Fukushima, T.; Fukuyama, T.; Tanaka, Y.; Sawasaki, T.; et al. The ubiquitin ligase STUB1 regulates stability and activity of RUNX1 and RUNX1-RUNX1T1. J. Biol. Chem. 2017, 292, 12528–12541. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhou, J.; Zhou, P.; Chen, W.; Guo, F. The ubiquitin ligase CHIP inactivates NF-κB signaling and impairs the ability of migration and invasion in gastric cancer cells. Int. J. Oncol. 2015, 46, 2096–2106. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, L.; He, X.; Shen, Y.; Liu, X.; Wei, J.; Yu, F.; Tian, J. CHIP promotes thyroid cancer proliferation via activation of the MAPK and AKT pathways. Biochem. Biophys. Res. Commun. 2016, 477, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, L.; Liang, Z.Y.; Jin, K.M.; Zhou, W.X.; Xing, B.C. Clinicopathologic and Prognostic Significance of Carboxyl Terminus of Hsp70-interacting Protein in HBV-related Hepatocellular Carcinoma. Asian Pac. J. Cancer Prev. 2015, 16, 3709–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravalin, M.; Theofilas, P.; Basu, K.; Opoku-Nsiah, K.A.; Assimon, V.A.; Medina-Cleghorn, D.; Chen, Y.F.; Bohn, M.F.; Arkin, M.; Grinberg, L.T.; et al. Specificity for latent C termini links the E3 ubiquitin ligase CHIP to caspases. Nat. Chem. Biol. 2019, 15, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.; Ye, F.; He, X.-X. The role of YWHAZ in cancer: A maze of opportunities and challenges. J. Cancer 2020, 11, 2252–2264. [Google Scholar] [CrossRef]
- Li, T.; Paudel, H.K. 14-3-3ζ Facilitates GSK3β-catalyzed tau phosphorylation in HEK-293 cells by a mechanism that requires phosphorylation of GSK3β on Ser9. Neurosci. Lett. 2007, 414, 203–208. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Han, D.; MacDonald, R.; Paudel, H.K. Overexpression of 14-3-3z promotes tau phosphorylation at Ser262 and accelerates proteosomal degradation of synaptophysin in rat primary hippocampal neurons. PLoS ONE 2013, 8, e84615. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, H.Y.; Li, T.; MacDonald, R.; Cho, C.M.; Leclerc, N.; Paudel, H.K. Interaction of 14-3-3ζ with microtubule-associated protein tau within Alzheimer’s disease neurofibrillary tangles. Biochemistry 2013, 52, 6445–6455. [Google Scholar] [CrossRef]
- Agarwal-Mawal, A.; Qureshi, H.Y.; Cafferty, P.W.; Yuan, Z.; Han, D.; Lin, R.; Paudel, H.K. 14-3-3 connects glycogen synthase kinase-3 beta to tau within a brain microtubule-associated tau phosphorylation complex. J. Biol. Chem. 2003, 278, 12722–12728. [Google Scholar] [CrossRef] [Green Version]
- Bustos, D.M. The role of protein disorder in the 14-3-3 interaction network. Mol. Biosyst. 2012, 8, 178–184. [Google Scholar] [CrossRef]
- Hernández, F.; Cuadros, R.; Avila, J. Zeta 14-3-3 protein favours the formation of human tau fibrillar polymers. Neurosci. Lett. 2004, 357, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Sadik, G.; Tanaka, T.; Kato, K.; Yamamori, H.; Nessa, B.N.; Morihara, T.; Takeda, M. Phosphorylation of tau at Ser214 mediates its interaction with 14-3-3 protein: Implications for the mechanism of tau aggregation. J. Neurochem. 2009, 108, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, M.; Sobue, K.; Paudel, H.K. 14-3-3ζ Is an Effector of Tau Protein Phosphorylation. J. Biol. Chem. 2000, 275, 25247–25254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umahara, T.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Iwamoto, T.; Ikeda, K.; Takasaki, M. 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer’s disease. Acta Neuropathol. 2004, 108, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Umahara, T.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Iwamoto, T.; Takasaki, M. Immunolocalization of 14-3-3 isoforms in brains with Pick body disease. Neurosci. Lett. 2004, 371, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lubec, G.; Nonaka, M.; Krapfenbauer, K.; Gratzer, M.; Cairns, N.; Fountoulakis, M. Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down syndrome and Alzheimer’s disease brain is downregulated at the mRNA and dysregulated at the protein level. J. Neural Transm. Suppl. 1999, 57, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Holzmann, C.; Riess, O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003, 4, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Papanikolopoulou, K.; Grammenoudi, S.; Samiotaki, M.; Skoulakis, E.M.C. Differential effects of 14-3-3 dimers on Tau phosphorylation, stability and toxicity in vivo. Hum. Mol. Genet. 2018, 27, 2244–2261. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, Y.-R.; Yang, H.-Y.; Li, X.-Z.; Jie, M.-M.; Hu, C.-J.; Wu, Y.-Y.; Yang, S.-M.; Yang, Y.-B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef]
- Driver, J.A.; Lu, K.P. Pin1: A new genetic link between Alzheimer’s disease, cancer and aging. Curr. Aging Sci. 2010, 3, 158–165. [Google Scholar] [CrossRef]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem. Sci. 2004, 29, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Dickson, D.W.; Davies, P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiol. Dis. 2003, 14, 251–264. [Google Scholar] [CrossRef]
- Lim, J.; Balastik, M.; Lee, T.H.; Nakamura, K.; Liou, Y.C.; Sun, A.; Finn, G.; Pastorino, L.; Lee, V.M.; Lu, K.P. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Investig. 2008, 118, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Poppek, D.; Keck, S.; Ermak, G.; Jung, T.; Stolzing, A.; Ullrich, O.; Davies, K.J.; Grune, T. Phosphorylation inhibits turnover of the tau protein by the proteasome: Influence of RCAN1 and oxidative stress. Biochem. J. 2006, 400, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhou, Y.; Chen, D.; Lee, T.H. Peptidyl-Prolyl Cis/Trans Isomerase Pin1 and Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 8, 355. [Google Scholar] [CrossRef]
- Kutter, S.; Eichner, T.; Deaconescu, A.M.; Kern, D. Regulation of Microtubule Assembly by Tau and not by Pin1. J. Mol. Biol. 2016, 428, 1742–1759. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, J.; Jung, E.S.; Kim, M.-H.; Kim, I.B.; Son, H.; Kim, S.; Kim, S.; Park, Y.M.; Mook-Jung, I.; et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun. 2019, 10, 3090. [Google Scholar] [CrossRef]
- Eichner, T.; Kutter, S.; Labeikovsky, W.; Buosi, V.; Kern, D. Molecular Mechanism of Pin1-Tau Recognition and Catalysis. J. Mol. Biol. 2016, 428, 1760–1775. [Google Scholar] [CrossRef]
- Smet, C.; Wieruszeski, J.M.; Buée, L.; Landrieu, I.; Lippens, G. Regulation of Pin1 peptidyl-prolyl cis/trans isomerase activity by its WW binding module on a multi-phosphorylated peptide of Tau protein. FEBS Lett. 2005, 579, 4159–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, P.; Cantrelle, F.-X.; Huvent, I.; Hanoulle, X.; Lopez, J.; Smet, C.; Wieruszeski, J.-M.; Landrieu, I.; Lippens, G. Proline Conformation in a Functional Tau Fragment. J. Mol. Biol. 2016, 428, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.H.; Tu, C.; Cao, W.; Klein, A.; Ramsey, R.; Fennell, B.J.; Lambert, M.; Ní Shúilleabháin, D.; Autin, B.; Kouranova, E.; et al. An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition. J. Biol. Chem. 2012, 287, 44425–44434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Liu, H.; Gu, S.; Wei, Z.; Liu, H. The role of Capon in multiple myeloma. Tumour Biol. 2017, 39, 1010428317713674. [Google Scholar] [CrossRef] [Green Version]
- Fuller-Pace, F.V. DEAD box RNA helicase functions in cancer. RNA Biol. 2013, 10, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Sen, B.; Johnson, F.M. Regulation of SRC family kinases in human cancers. J. Signal Transduct. 2011, 2011, 865819. [Google Scholar] [CrossRef] [Green Version]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, A.; Nanjappa, V.; Raja, R.; Sathe, G.; Puttamallesh, V.N.; Jain, A.P.; Pinto, S.M.; Balaji, S.A.; Chavan, S.; Sahasrabuddhe, N.A.; et al. A dual specificity kinase, DYRK1A, as a potential therapeutic target for head and neck squamous cell carcinoma. Sci. Rep. 2016, 6, 36132. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P. Ewing Sarcoma Protein: A Key Player in Human Cancer. Int. J. Cell Biol. 2013, 2013, 642853. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papin, S.; Paganetti, P. Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sci. 2020, 10, 862. https://doi.org/10.3390/brainsci10110862
Papin S, Paganetti P. Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sciences. 2020; 10(11):862. https://doi.org/10.3390/brainsci10110862
Chicago/Turabian StylePapin, Stéphanie, and Paolo Paganetti. 2020. "Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer" Brain Sciences 10, no. 11: 862. https://doi.org/10.3390/brainsci10110862
APA StylePapin, S., & Paganetti, P. (2020). Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sciences, 10(11), 862. https://doi.org/10.3390/brainsci10110862