Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Preparation
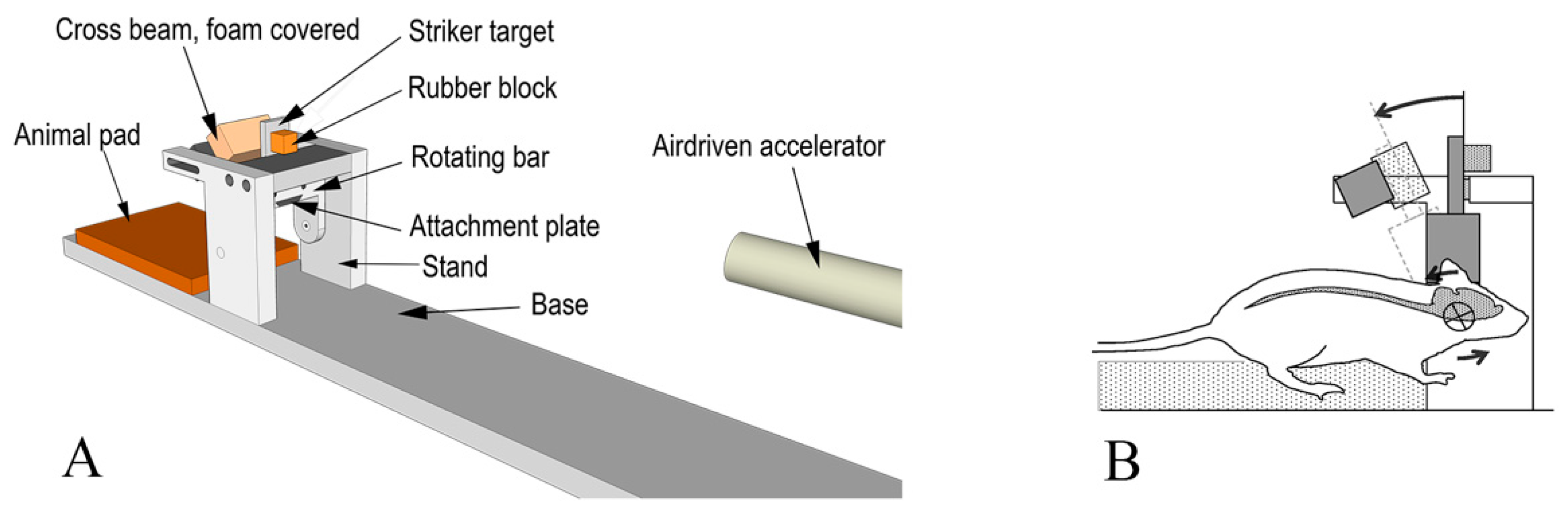
2.2. Experimental Setup
2.3. Dissection and Immunohistochemistry
2.4. RNAscope® ISH
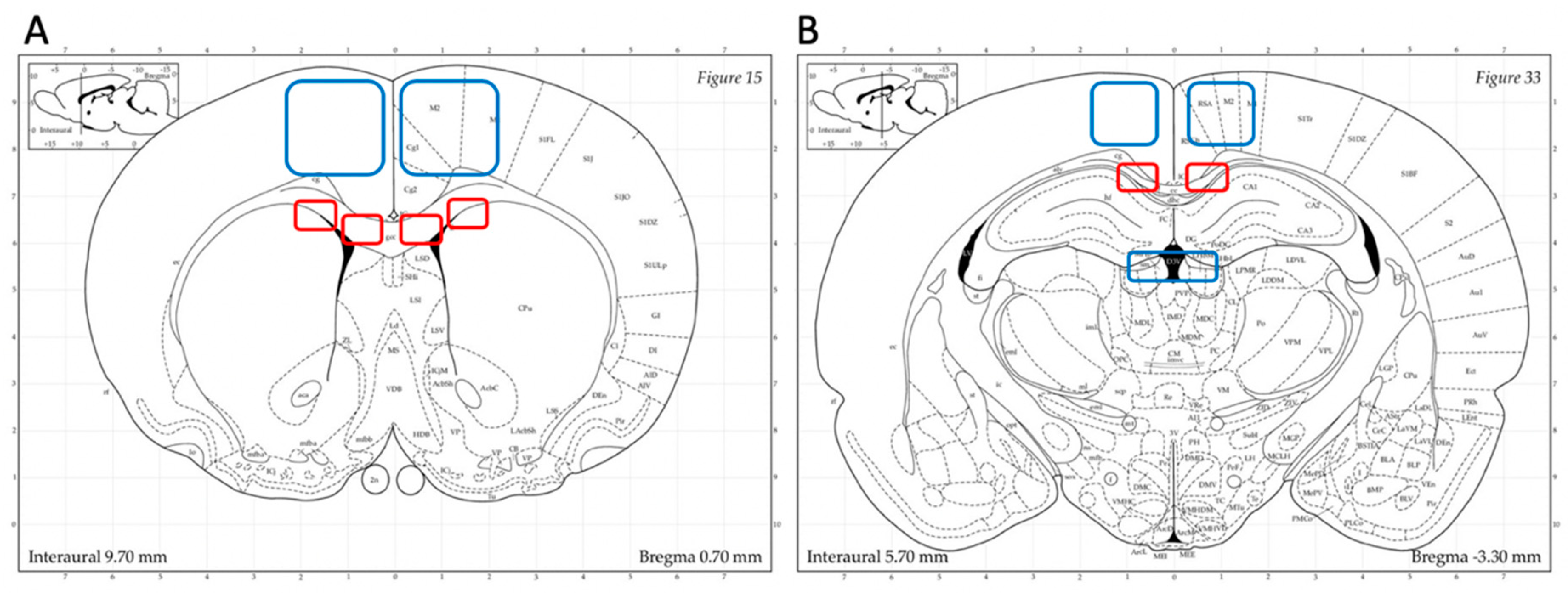
2.5. Microscopic and Statistical Analyses
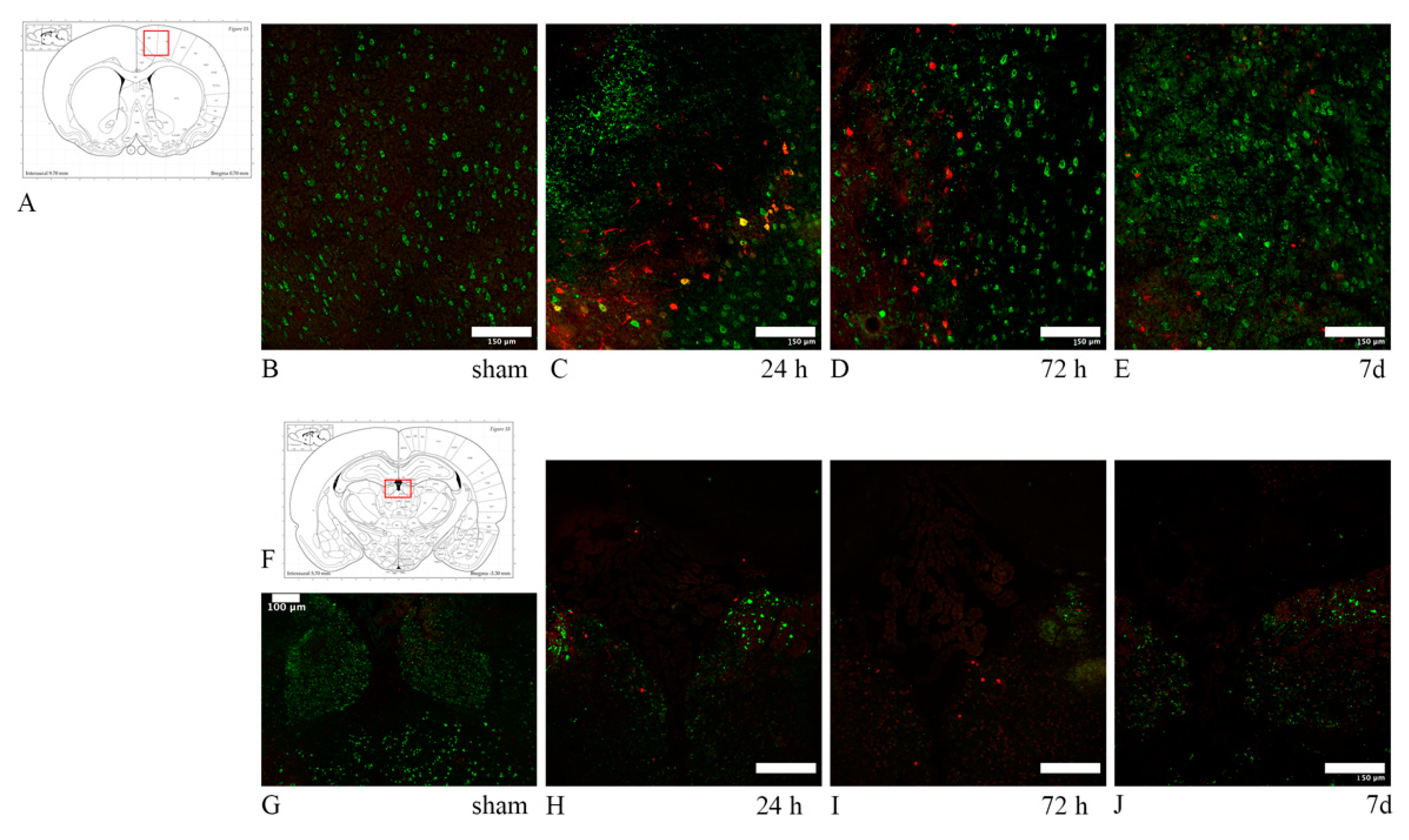
3. Results
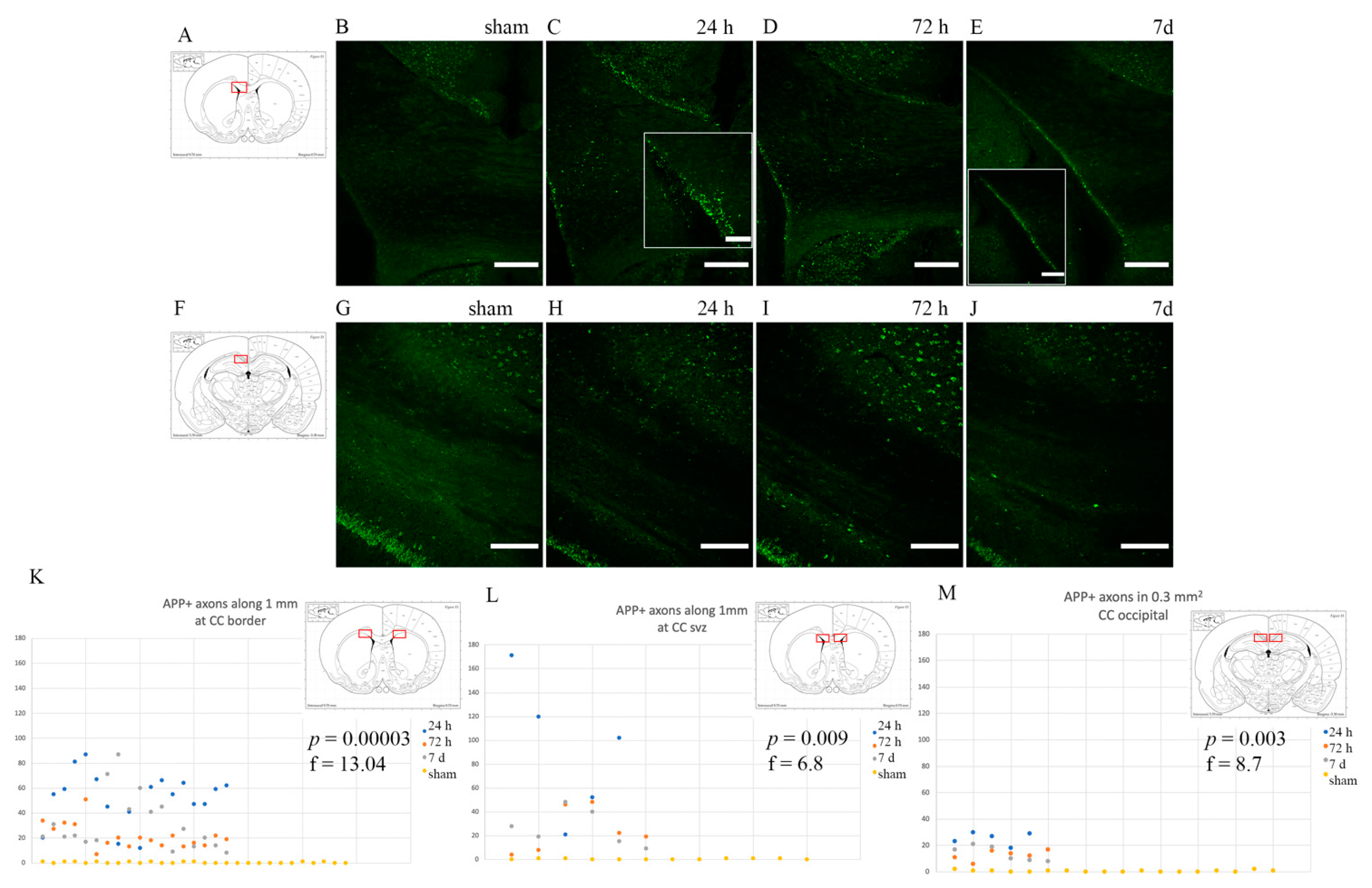
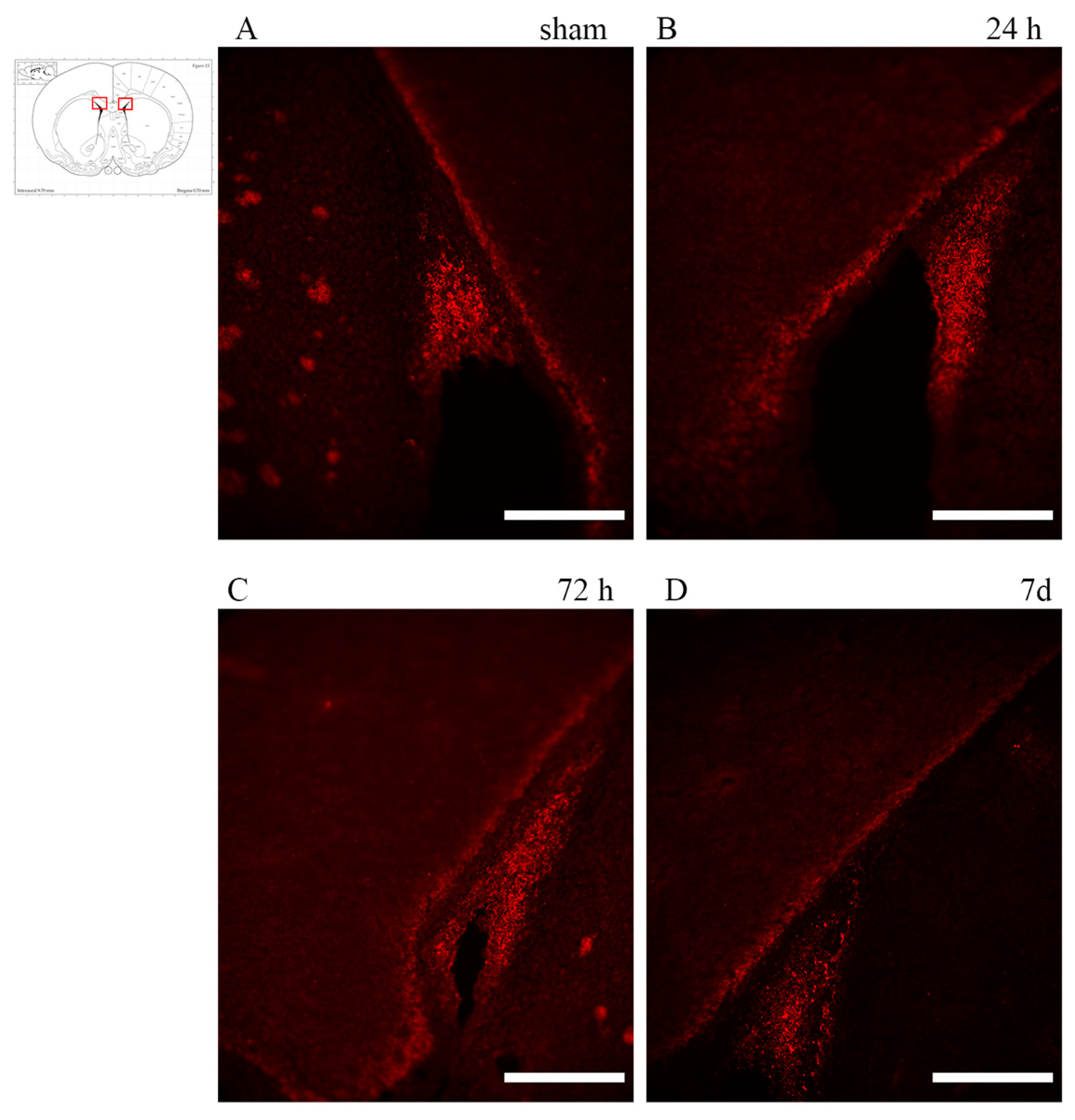
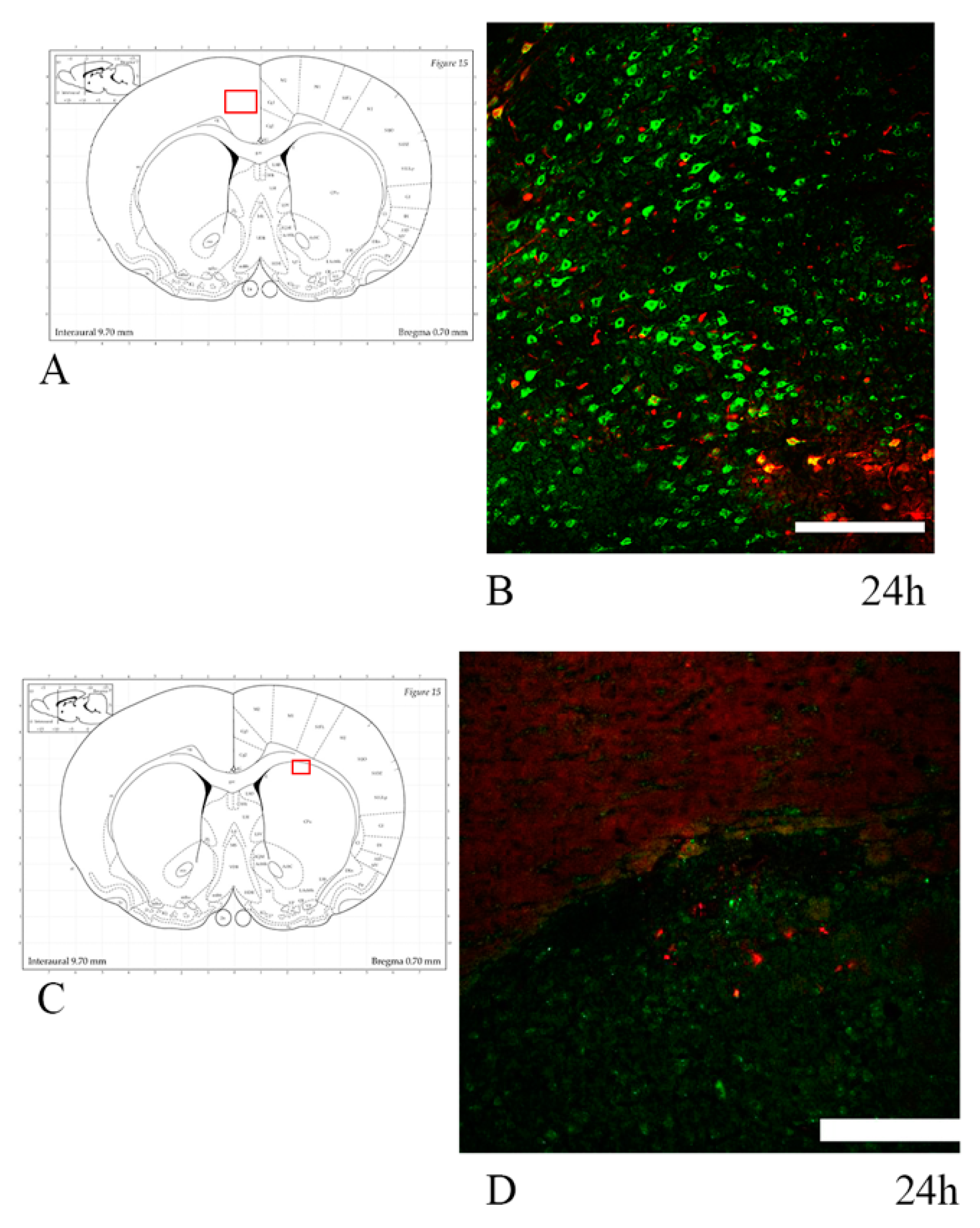
3.1. Axonal Injury: Anti-APP Antibody
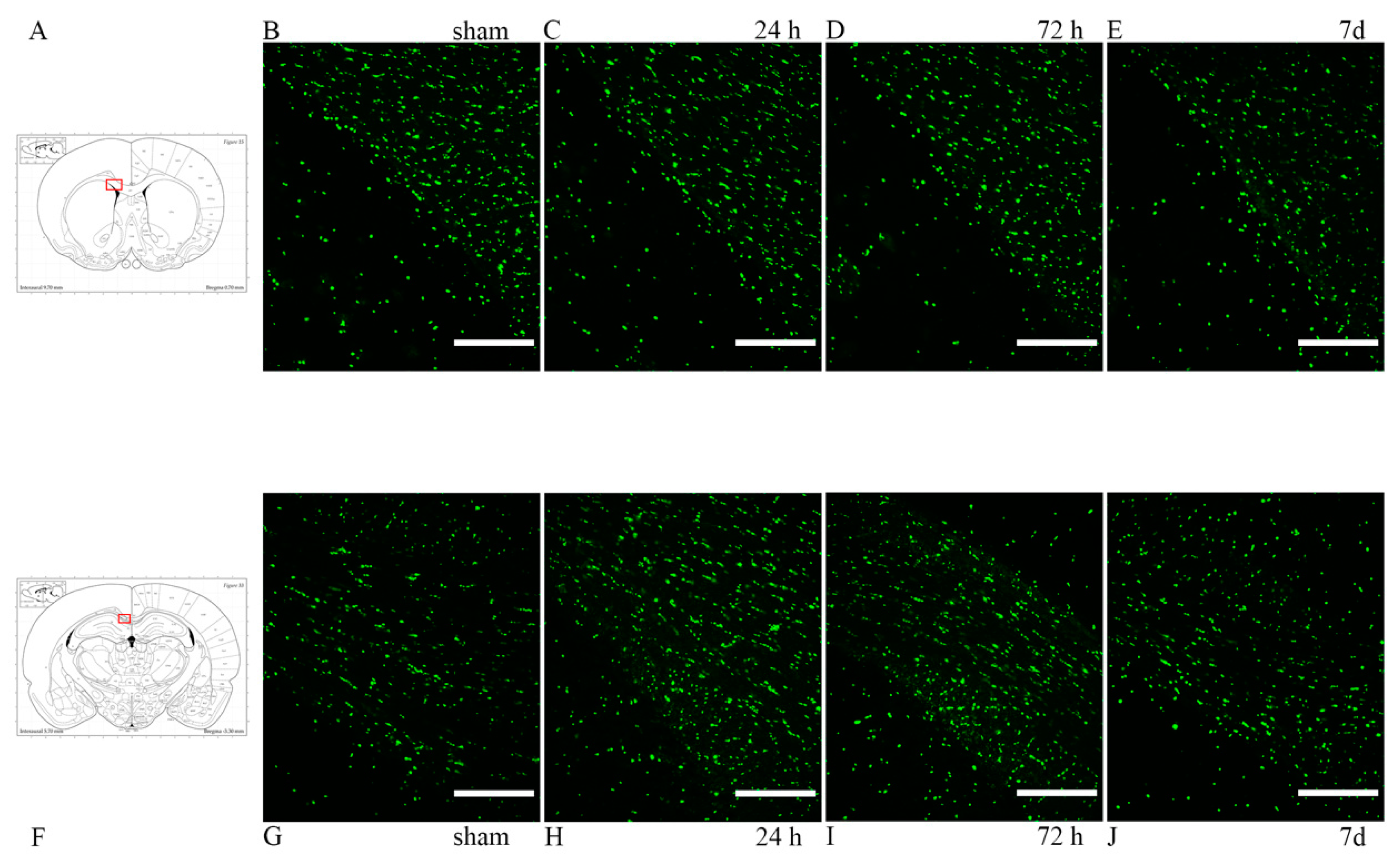
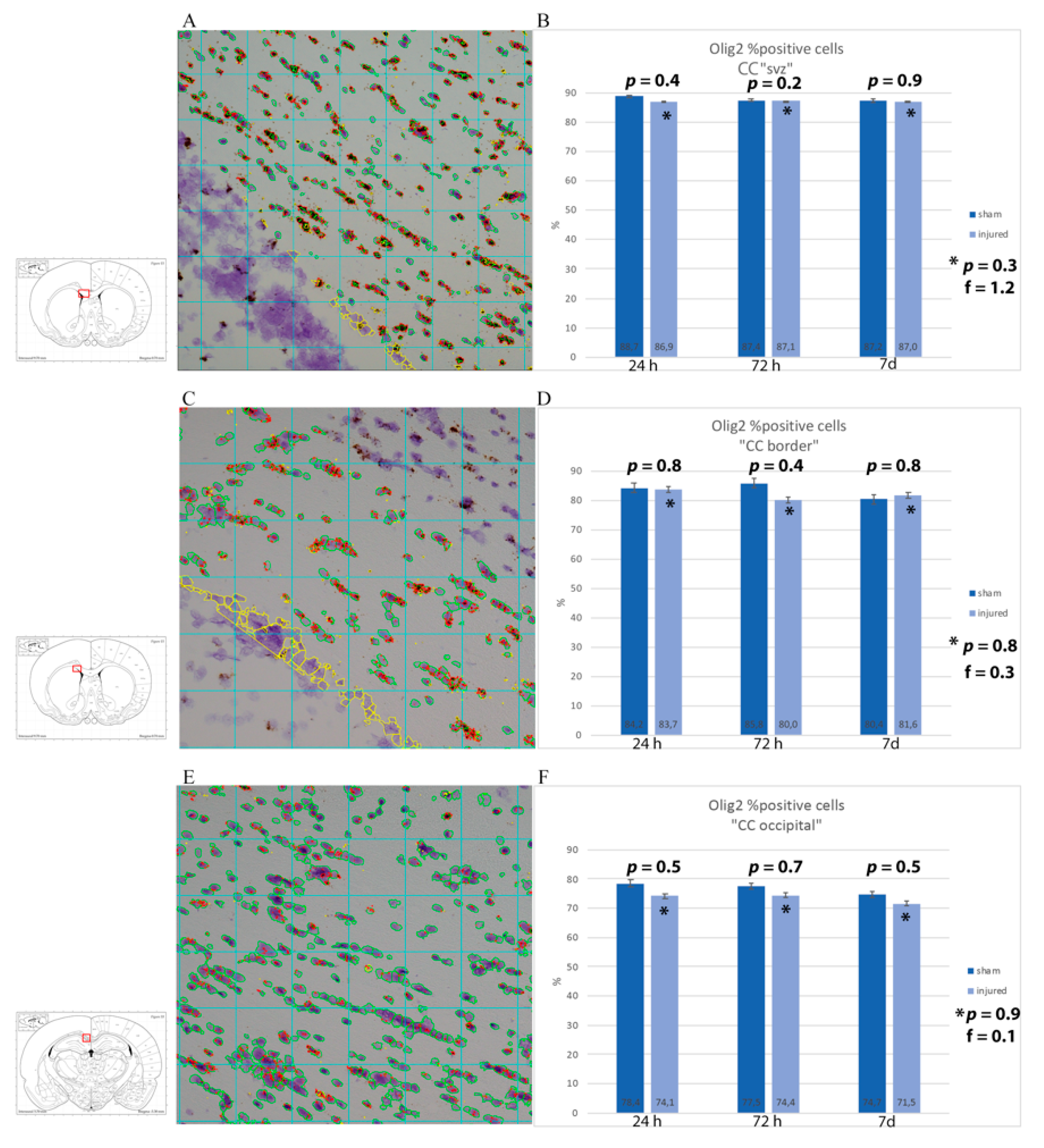
3.2. Oligodendrocyte Lineage Cells: Anti-Olig2 Antibody and Olig2 mRNA
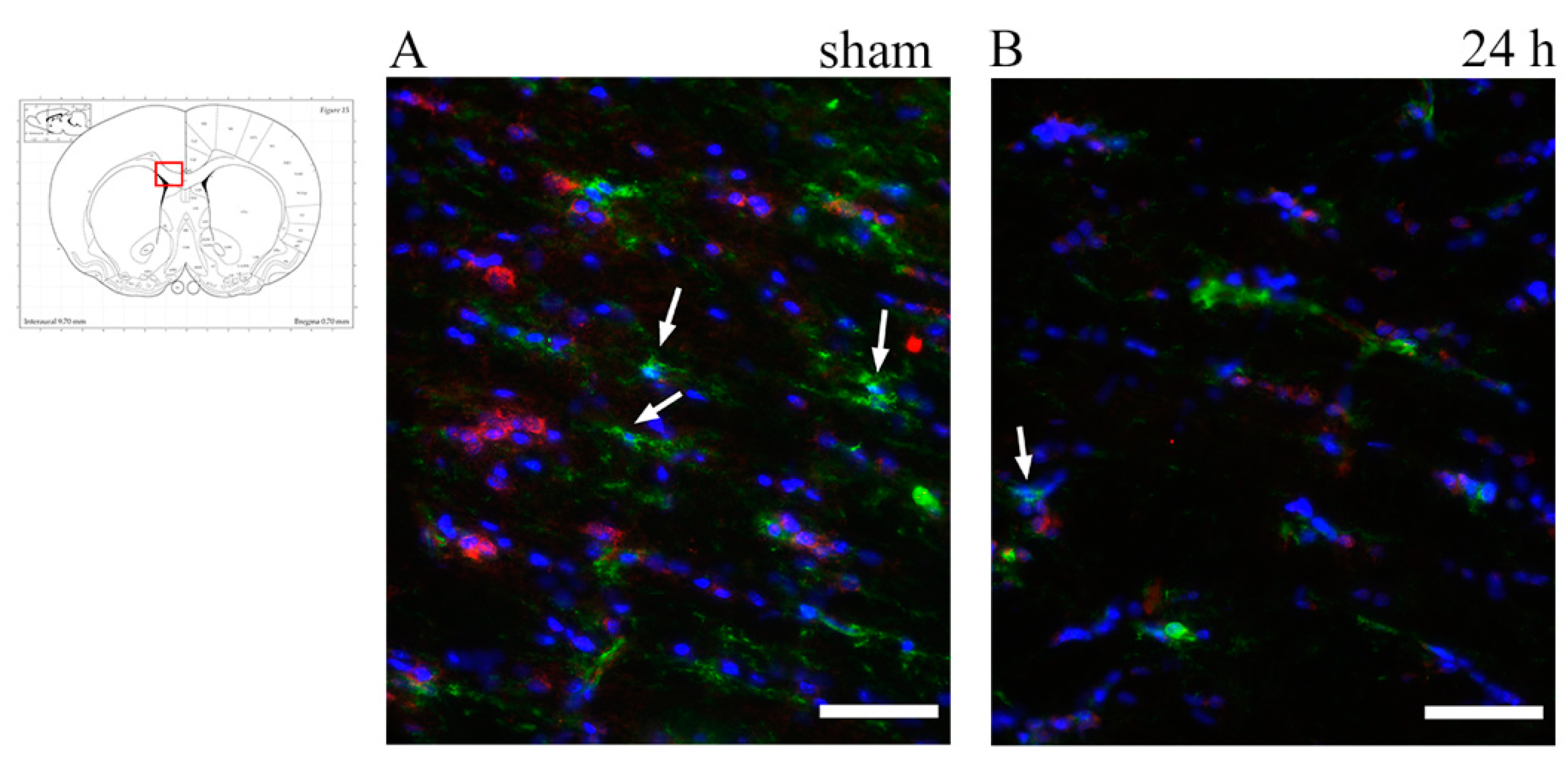
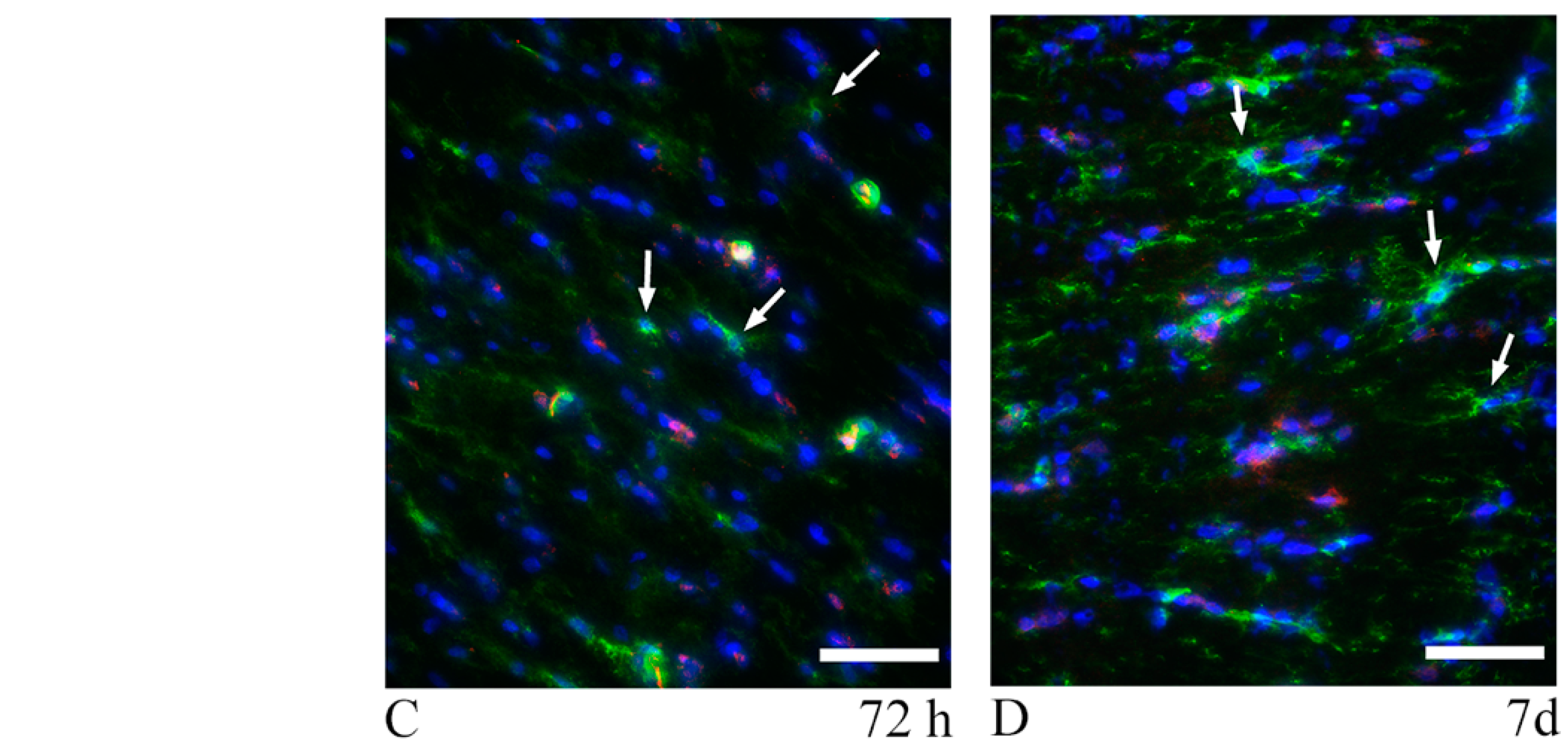
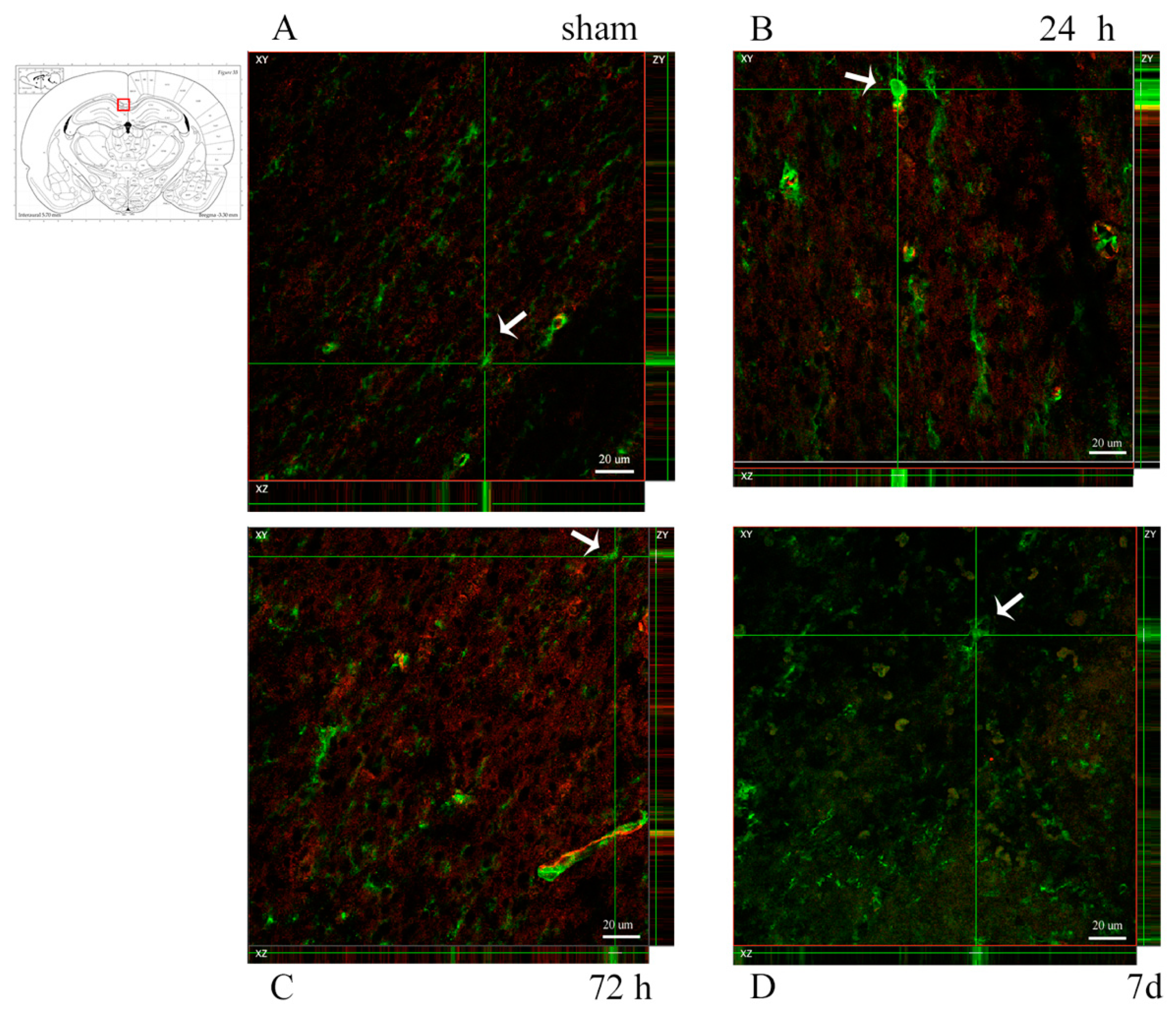
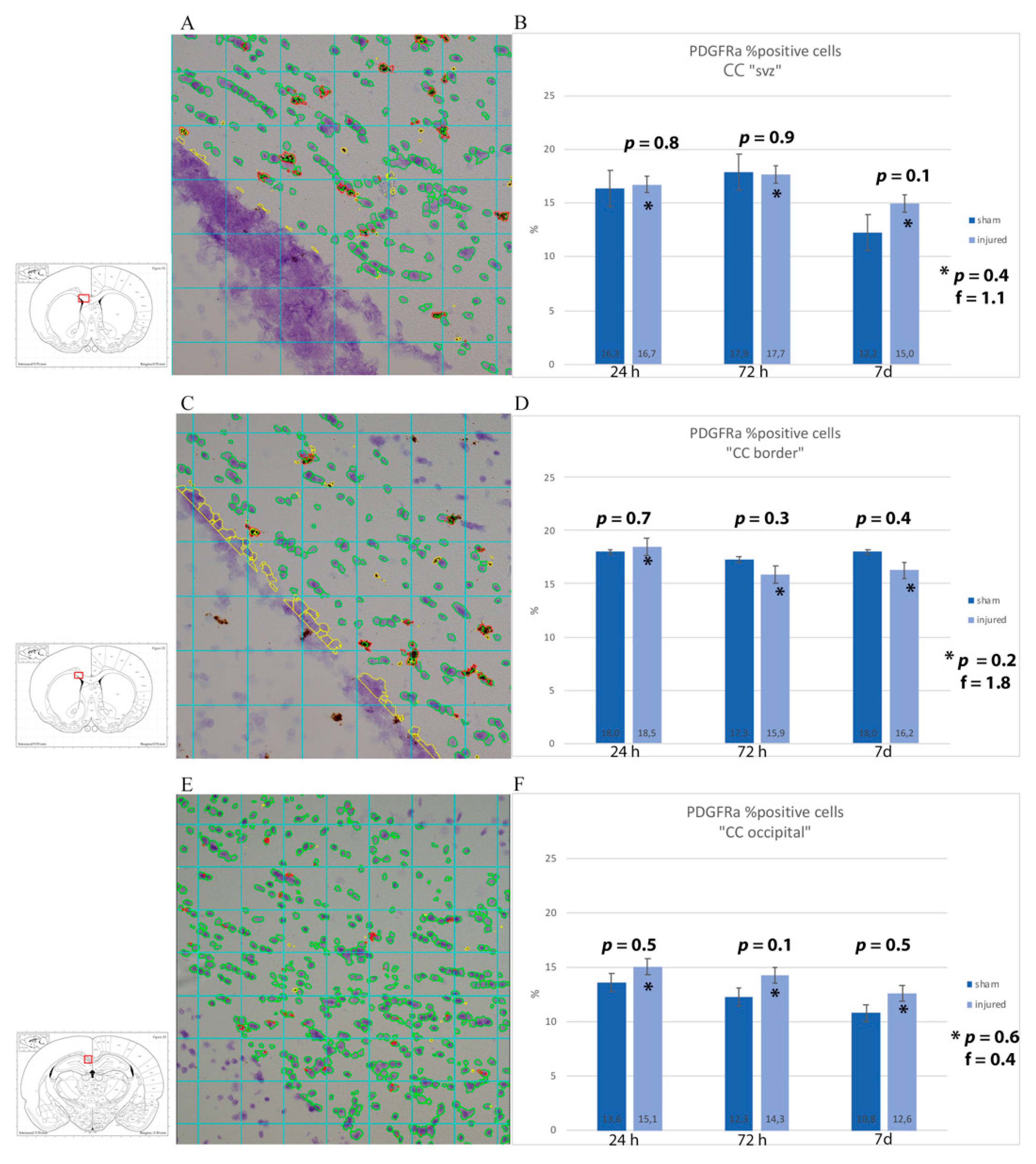
3.3. Oligodendrocyte Progenitor Cells: Anti-NG2, Anti-A2B5 Antibodies and PDGFRa mRNA
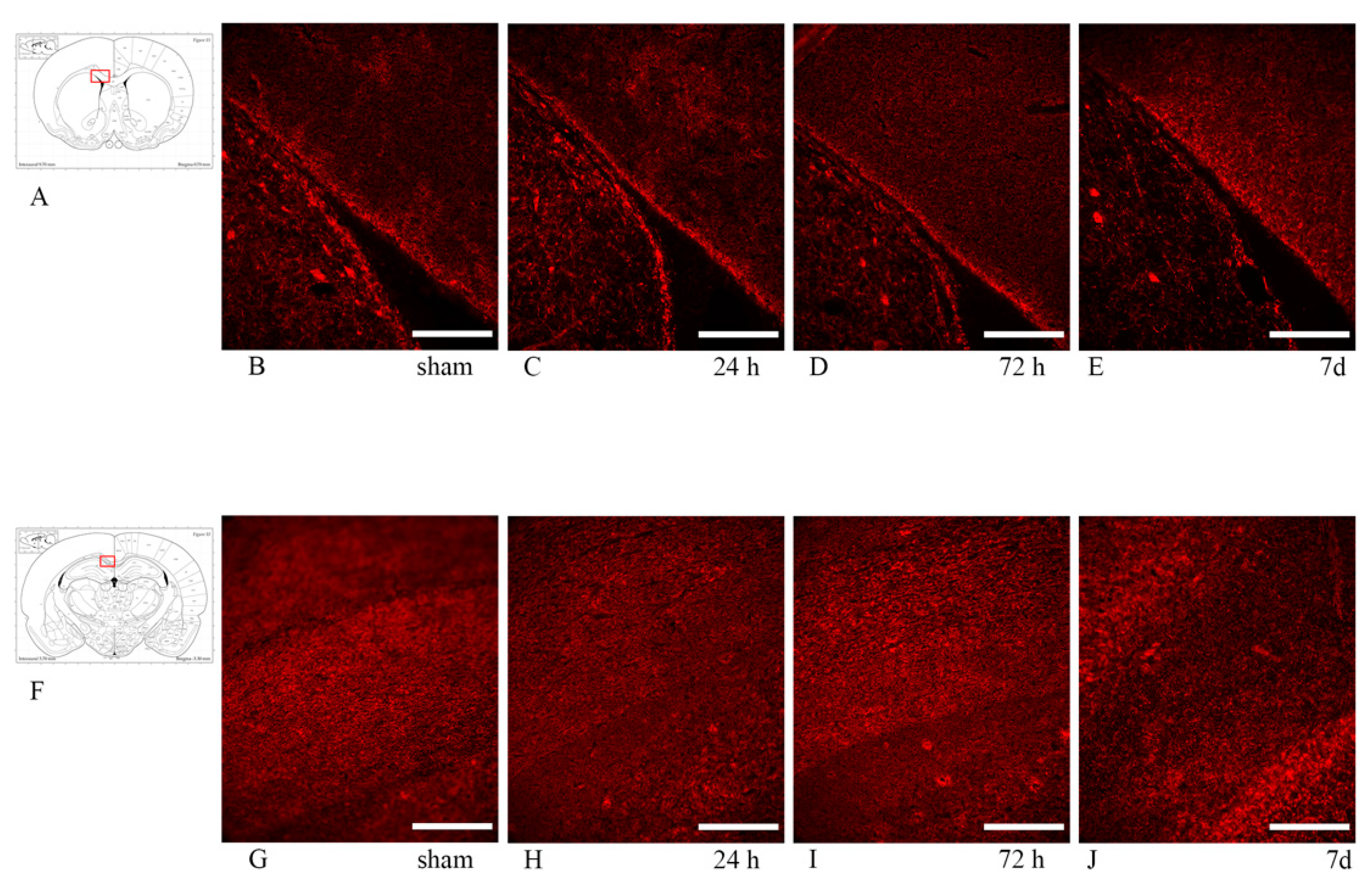
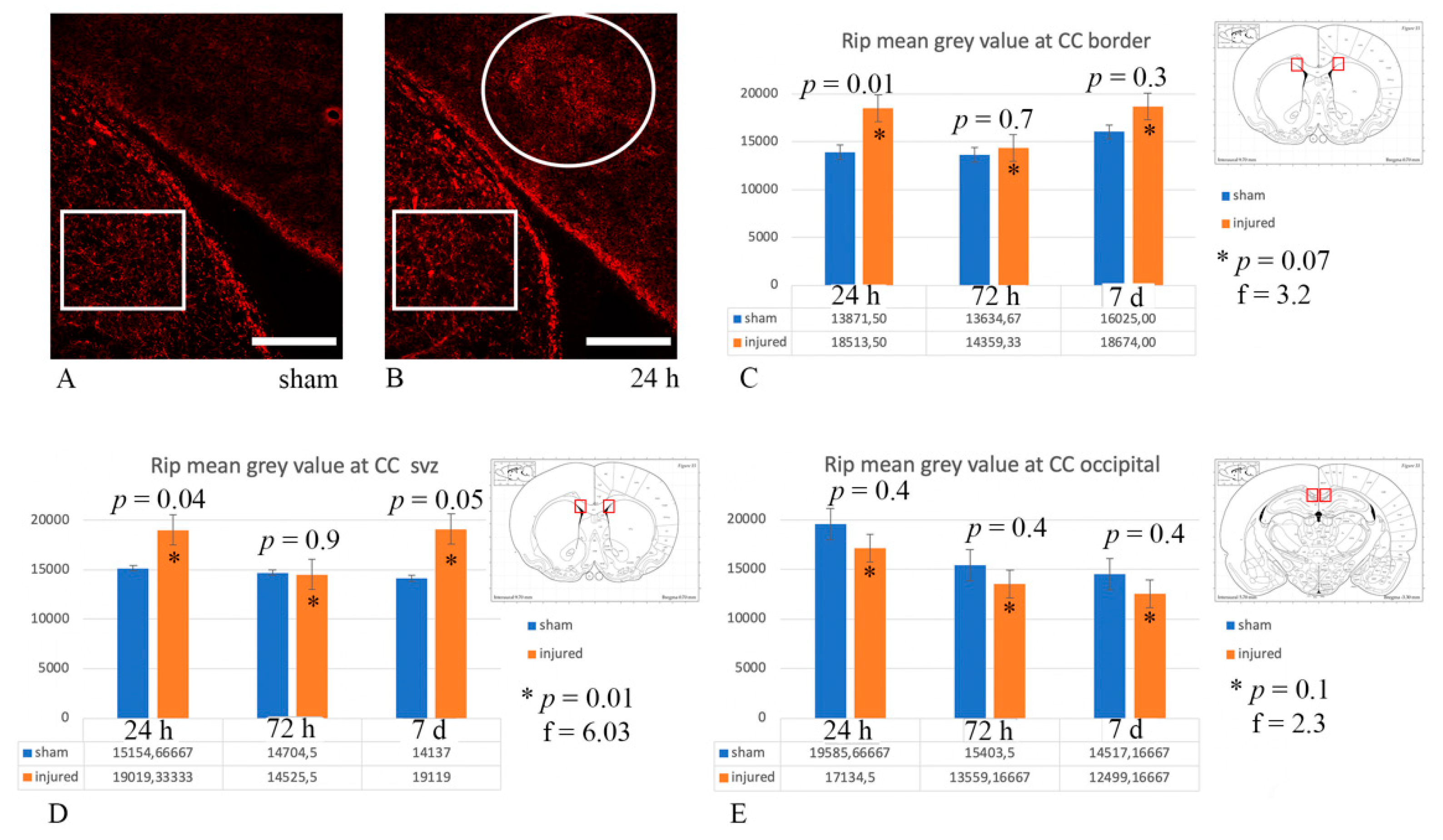
3.4. Myelin Pathology: Anti-MBP and Anti-Rip Antibodies
3.5. Neuronal Stress: Anti-HSP70 and Anti-ATF3 Antibodies
4. Discussion
4.1. Axonal Injury in TBI
4.2. Oligodendrocyte Loss
4.3. Myelin Injury
4.4. TBI-Induced OPCs Reaction
4.5. Neuronal Somata Stress
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Majdan, M.; Plancikova, D.; Brazinova, A.; Rusnak, M.; Nieboer, D.; Feigin, V.; Maas, A. Epidemiology of traumatic brain injuries in Europe: A cross-sectional analysis. Lancet Public Health 2016, 1, e76–e83. [Google Scholar] [CrossRef]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef] [Green Version]
- Holbourn, A.H.S. The mechanics of brain injuries. Br. Med. Bull. 1945, 3, 147–149. [Google Scholar] [CrossRef]
- Meythaler, J.M.; Peduzzi, J.D.; Eleftheriou, E.; Novack, T.A. Current concepts: Diffuse axonal injury-associated traumatic brain injury. Arch. Phys. Med. Rehabil. 2001, 82, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- McAllister, T.W. Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 2011, 13, 287–300. [Google Scholar]
- Povlishock, J.T.; Christman, C.W. The pathology of traumatically induced axonal injury in animals and humans: A review of current thoughts. J. Neurotrauma 1995, 12, 555–564. [Google Scholar] [CrossRef]
- Gennarelli, T.A.; Thibault, L.E.; Graham, D.I. Diffuse axonal injury: An important form of traumatic brain damage. Neuroscience 1998, 4, 202–215. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Katz, D.I. Update of neuropathology and neurological recovery after traumatic brain injury. J. Head Trauma Rehabil. 2005, 20, 76–94. [Google Scholar] [CrossRef] [Green Version]
- Büki, A.; Povlishock, J.T. All roads lead to disconnection? Traumatic axonal injury revisited. Acta Neurochir. 2006, 148, 181–193. [Google Scholar] [CrossRef]
- Tehse, J.; Taghibiglou, C. The overlooked aspect of excitotoxicity: Glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. 2019, 49, 1157–1170. [Google Scholar] [CrossRef]
- Smith, D.H.; Meaney, D.F.; Shull, W.H. Diffuse axonal injury in head trauma. J. Head Trauma Rehabil. 2003, 18, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, S.M.; Nash, M.J.; Sweeting, C.J.; Graham, D.I.; Roberts, G.W. β-amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci. Lett. 1993, 160, 139–144. [Google Scholar] [CrossRef]
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol. Res. 2009, 31, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Singleton, R.H. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: Evidence for multiple independent injury phenotypes. J. Neurosci. 2004, 24, 3543–3553. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Smith, D.H.; Meaney, D.F. In-vitro approaches for studying blast-induced traumatic brain injury. J. Neurotrauma 2009, 26, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Hu, B.; Alonso, O.F.; Bramlett, H.M.; Kokame, K.; Dietrich, W.D. Subcellular stress response after traumatic brain injury. J. Neurotrauma 2007, 24, 599–612. [Google Scholar] [CrossRef]
- Rajdev, S.; Sharp, F.R. Stress proteins as molecular markers of neurotoxicity. Toxicol. Pathol. 2000, 28, 105–112. [Google Scholar] [CrossRef] [Green Version]
- DeGracia, D.J.; Kreipke, C.W.; Kayali, F.M.; Rafols, J.A. Brain endothelial HSP-70 stress response coincides with endothelial and pericyte death after brain trauma. Neurol. Res. 2007, 29, 356–361. [Google Scholar] [CrossRef]
- Chen, M.; Clark, R.S.B.; Kochanek, P.M.; Chen, J.; Schiding, J.K.; Stetler, R.A.; Simon, R.P.; Graham, S.H. 72-kDa heat shock protein and mRNA expression after controlled cortical impact injury with hypoxemia in rats. J. Neurotrauma 1998, 15, 171–181. [Google Scholar] [CrossRef]
- Armstrong, R.C.; Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp. Neurol. 2016, 275, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.C.; Akle, V.; Zheng, W.; Kitlen, J.; O’Steen, B.; Larner, S.F.; Dave, J.R.; Tortella, F.C.; Hayes, R.L.; Wang, K.K.W. Extensive degradation of myelin basic protein isoforms by calpain following traumatic brain injury. J. Neurochem. 2006, 98, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Ottens, A.K.; Golden, E.C.; Bustamante, L.; Hayes, R.L.; Denslow, N.D.; Wang, K.K.W. Proteolysis of multiple myelin basic protein isoforms after neurotrauma: Characterization by mass spectrometry. J. Neurochem. 2008, 104, 1404–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saatman, K.E.; Creed, J.; Raghupathi, R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 2010, 7, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Truong, L.; Bear, D.; Chafik, D.; Modafferi, E.; Hung, C.T. Shear stress alters the expression of myelin-associated glycoprotein (MAG) and myelin basic protein (MBP) in Schwann cells. J. Orthop. Res. 2005, 23, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.M.; Mierzwa, A.J.; Kijpaisalratana, N.; Tang, H.; Wang, Y.; Song, S.K.; Selwyn, R.; Armstrong, R.C. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the Corpus callosum. J. Neuropathol. Exp. Neurol. 2013, 72, 1106–1125. [Google Scholar] [CrossRef] [Green Version]
- Tyurin, V.; Tyurina, Y.Y.; Borisenko, G.G.; Sokolova, T.V.; Ritov, V.B.; Quinn, P.J.; Rose, M.; Kochanek, P.; Graham, S.H.; Kagan, V.E. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000, 75, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Merrill, J.E.; Scolding, N.J. Mechanisms of damage to myelin and oligodendrocytes and their relevance to disease. Neuropathol. Appl. Neurobiol. 1999, 25, 435–458. [Google Scholar] [CrossRef]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Swedlow, P.E.; Styren, S.D.; DeKosky, S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993, 61, 2015–2024. [Google Scholar] [CrossRef]
- Kou, Z.; Vande Vord, P.J. Traumatic white matter injury and glial activation: From basic science to clinics. Glia 2014, 62, 1831–1855. [Google Scholar] [CrossRef]
- Caprariello, A.; Mangla, S.; Miller, R.H.; Selkirk, S.M. Apoptosis of oligodendroytes in the CNS results in rapid focal demyelination. Ann. Neurol. 2012, 72, 395–405. [Google Scholar] [CrossRef]
- Lotocki, G.; de Vaccari, J.R.; Alonso, O.; Molano, J.S.; Nixon, R.; Safavi, P.; Dietrich, W.D.; Bramlett, H.M. Oligodendrocyte vulnerability following traumatic brain injury in rats. Neurosci. Lett. 2011, 499, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.C.; Mierzwa, A.J.; Sullivan, G.M.; Sanchez, M.A. Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 2016, 110, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flygt, J.; Djupsjö, A.; Lenne, F.; Marklund, N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur. J. Neurosci. 2013, 38, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Dent, K.A.; Christie, K.J.; Bye, N.; Basrai, H.S.; Turbic, A.; Habgood, M.; Cate, H.S.; Turnley, A.M. Oligodendrocyte birth and death following traumatic brain injury in adult mice. PLoS ONE 2015, 10, e0121541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, M. Expression of transcription factors during oligodendroglial development. Microsc. Res. Tech. 2001, 52, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Chang, A.; Trapp, B.D. NG2+ glial cells: A novel glia cell population in the adult brain. J. Neuropathol. Exp. Neurol. 1999, 58, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- McTigue, D.M.; Tripathi, R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef]
- Levine, J.M.; Reynolds, R.; Fawcett, J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Scheller, A.; Bai, X.; Kirchhoff, F. The role of the oligodendrocyte lineage in acute brain trauma. Neurochem. Res. 2017, 42, 1–11. [Google Scholar] [CrossRef]
- Flygt, J.; Clausen, F.; Marklund, N. Diffuse traumatic brain injury in the mouse induces a transient proliferation of oligodendrocyte progenitor cells in injured white matter tracts. Restor. Neurol. Neurosci. 2017, 35, 251–263. [Google Scholar] [CrossRef]
- Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M.; McDaniel, D.P.; Armstrong, R.C. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 2015, 74, 218–232. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.J.M.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.L.; Yamaguchi, Y.; Stallcup, W.B.; Tuszynski, M.H. NG2 is a major chondroitin sulfate Proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J. Neurosci. 2002, 22, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J. Neurosci. 1994, 14, 4716–4730. [Google Scholar] [CrossRef]
- Davidsson, J.; Risling, M. A new model to produce sagittal plane rotational induced diffuse axonal injuries. Front. Neurol. 2011, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagnostics 2012, 14, 22–29. [Google Scholar] [CrossRef] [Green Version]
- ACD, Using ImageJ to Analyze RNAscope® and BaseScope TM Data Workflow. Technical Note. Available online: https://acdbio.com. (accessed on 4 November 2019).
- Hardy, W.N.; Mason, M.J.; Foster, C.D.; Shah, C.S.; Kopacz, J.M.; Yang, H.; King, A.I.; Bishop, J.; Bey, M.; Yang, K.H.; et al. A study of the response of the human cadaver head to impact. Stapp Car Crash J. 2007, 51, 17–80. [Google Scholar]
- Baumgartner, D.; Lamy, M.; Willinger, R.; Choquet, P.; Goetz, C.; Constantinesco, A.; Davidsson, J. Finite element analysis of traumatic brain injuries mechanisms in the rat. In Proceedings of the 2009 IRCOBI Conference, York, UK, 9–11 September 2009. [Google Scholar]
- Clayton, E.H.; Genin, G.M.; Bayly, P.V. Transmission, attenuation and reflection of shear waves in the human brain. J. R. Soc. Interface 2012, 9, 2899–2910. [Google Scholar] [CrossRef]
- Chafi, M.S.; Karami, G.; Ziejewski, M. Biomechanical assessment of brain dynamic responses due to blast pressure waves. Ann. Biomed. Eng. 2010, 38, 490–504. [Google Scholar] [CrossRef]
- Smith, D.H.; Meaney, D.F. Axonal Damage in Traumatic Brain Injury. Neurosci. 2000, 6, 483–495. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, K.; Wang, Z.; Chen, G. Progress of research on diffuse axonal injury after traumatic brain injury. Neural Plast. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambri, M.; Djurovic, V.; Kibble, M.; Cairns, N.; Al-Sarraj, S. Specificity and sensitivity of βAPP in head injury. Clin. Neuropathol. 2001, 20, 263–271. [Google Scholar] [PubMed]
- Riise, J.; Pakkenberg, B. Stereological estimation of the total number of myelinated callosal fibers in human subjects. J. Anat. 2011, 218, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Antona-Makoshi, J.; Eliasson, E.; Davidsson, J.; Ejima, S.; Ono, K. Effect of aging on brain injury prediction in rotational head trauma—A parameter study with a rat finite element model. Traffic Inj. Prev. 2015, 16, S91–S99. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Li, J.; Feng, D.F.; Gu, L. Diffuse axonal injury induced by simultaneous moderate linear and angular head accelerations in rats. Neuroscience 2010, 169, 357–369. [Google Scholar] [CrossRef]
- Wang, H.-C.; Duan, Z.-X.; Wu, F.-F.; Xie, L.; Zhang, H.; Ma, Y.-B. A new rat model for diffuse axonal injury using a combination of linear acceleration and angular acceleration. J. Neurotrauma 2010, 27, 707–719. [Google Scholar] [CrossRef]
- Chen, K.; Gu, H.; Zhu, L.; Feng, D.F. A new model of repetitive traumatic brain injury in mice. Front. Neurosci. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Ekmark-Lewén, S.; Flygt, J.; Kiwanuka, O.; Meyerson, B.J.; Lewén, A.; Hillered, L.; Marklund, N. Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes. J. Neuroinflamm. 2013, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.R.; Okonkwo, D.O.; Singleton, R.H.; Mutlu, L.K.; Helm, G.A.; Povlishock, J.T. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid α peptide in traumatic axonal injury. J. Neurotrauma 2002, 19, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Siman, R.; Iwata, A.; Meaney, D.F.; Trojanowski, J.Q.; Smith, D.H. Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004, 165, 357–371. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Kraydieh, S.; Green, E.J.; Dietrich, W.D. Temporal and regional patterns of axonal damage following traumatic brain injury: A β-amyloid precursor protein immunocytochemical study in rats. J. Neuropathol. Exp. Neurol. 1997, 56, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidsson, J.; Angeria, M.; Risling, M. Effect of age on amount and distribution of diffuse axonal injury after rotational trauma. In Proceedings of the JSAE Annual Congress, Yokohama, Japan, 24 May 2013. [Google Scholar]
- Conti, A.C.; Raghupathi, R.; Trojanowski, J.Q.; Mcintosh, T.K. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998, 18, 5663–5672. [Google Scholar] [CrossRef]
- Lotocki, G.; de Vaccari, J.R.; Alonso, O.; Molano, J.S.; Nixon, R.; Dietrich, W.D.; Bramlett, H.M. Oligodendrocyte vulnerability following traumatic brain injury in rats: Effect of moderate hypothermia. Ther. Hypothermia Temp. Manag. 2011, 1, 43–51. [Google Scholar] [CrossRef]
- Flygt, J.; Gumucio, A.; Ingelsson, M.; Skoglund, K.; Holm, J.; Alafuzoff, I.; Marklund, N. Human traumatic brain injury results in oligodendrocyte death and increases the number of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 2016, 75, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Haagsma, J.A.; Scholten, A.C.; Andriessen, T.M.J.C.; Vos, P.E.; van Beeck, E.F.; Polinder, S. Impact of depression and post-traumatic stress disorder on functional outcome and health-related quality of life of patients with mild traumatic brain injury. J. Neurotrauma 2015, 32, 853–862. [Google Scholar] [CrossRef]
- Ng, H.K.; Mahaliyana, R.D.; Poon, W.S. The pathological spectrum of diffuse axonal injury in blunt head trauma: Assessment with axon and myelin stains. Clin. Neurol. Neurosurg. 1994, 96, 24–31. [Google Scholar] [CrossRef]
- Taib, T.; Leconte, C.; van Steenwinckel, J.; Cho, A.H.; Palmier, B.; Torsello, E.; Kuen, R.L.; Onyeomah, S.; Ecomard, K.; Benedetto, C.; et al. Neuroinflammation, myelin and behavior: Temporal patterns following mild traumatic brain injury in mice. PLoS ONE 2017, 12, e0184811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhang, J.; Hu, X.; Zhang, L.; Mao, L.; Jiang, X.; Liou, A.K.F.; Leak, R.K.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1864–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Sakurai, Y.; Ichinose, T.; Aikawa, Y.; Kotani, M.; Itoh, K. Monoclonal antibody Rip specifically recognizes 2’,3’-cyclic nucleotide 3’-phosphodiesterase in oligodendrocytes. J. Neurosci. Res. 2006, 84, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Hockfield, S.; Black, J.A.; Woodruff, K.A.; Waxman, S.G. In situ demonstration of mature oligodendrocytes and their processes: An immunocytochemical study with a new monoclonal antibody, Rip. Glia 1989, 2, 380–390. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–28. [Google Scholar] [CrossRef]
- Fernandez-Castaneda, A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.S.; Mayer-Proschel, M. Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Dev. Biol. 1997, 188, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Babiarz, J.; Woodbury, J.; Kane-Goldsmith, N.; Grumet, M. Spatiotemporal heterogeneity of CNS radial glial cells and their transition to restricted precursors. Dev. Biol. 2004, 271, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Watanabe, M.; Yang, Z.; Bu, J. Identity, distribution, and development of polydendrocytes: NG2-expressing glial cells. J. Neurocytol. 2002, 31, 437–455. [Google Scholar] [CrossRef]
- Garcia-Verdugo, J.M.; Doetsch, F.; Wichterle, H.; Lim, D.A.; Alvarez-Buylla, A. Architecture and cell types of the adult subventricular zone: In search of the stem cells. J. Neurobiol. 1998, 36, 234–248. [Google Scholar] [CrossRef]
- Bye, N.; Carron, S.; Han, X.; Agyapomaa, D.; Ng, S.Y.; Yan, E.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J. Neurosci. Res. 2011, 89, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Macchi, M.; Cayre, M.; Durbec, P. Oligodendrogenesis in the normal and pathological central nervous system. Front. Neurosci. 2014, 8, 1–22. [Google Scholar]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [PubMed]
- Nait-Oumesmar, B.; Decker, L.; Lachapelle, F.; Avellana-Adalid, V.; Bachelin, C.; Baron-Van Evercooren, A. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur. J. Neurosci. 1999, 11, 4357–4366. [Google Scholar] [CrossRef]
- Dawson, M.R.L.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Mierzwa, A.J.; Sullivan, G.M.; Beer, L.A.; Ahn, S.; Armstrong, R.C. Comparison of cortical and white matter traumatic brain injury models reveals differential effects in the subventricular zone and divergent sonic hedgehog signaling pathways in neuroblasts and oligodendrocyte progenitors. ASN Neuro 2014, 6, 1–16. [Google Scholar] [CrossRef]
- Belachew, S.; Chittajallu, R.; Aguirre, A.A.; Yuan, X.; Kirby, M.; Anderson, S.; Gallo, V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J. Cell Biol. 2003, 161, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Moransard, M.; Dann, A.; Staszewski, O.; Fontana, A.; Prinz, M.; Suter, T. NG2 expressed by macrophages and oligodendrocyte precursor cells is dispensable in experimental autoimmune encephalomyelitis. Brain 2011, 134, 1315–1330. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Li, P.; Li, H.X.; Jiang, H.Y.; Zhu, L.; Wu, H.Y.; Li, J.T.; Lai, J.H. Expression of NG2 and platelet-derived growth factor receptor alpha in the developing neonatal rat brain. Neural Regen. Res. 2017, 12, 1843–1852. [Google Scholar] [CrossRef]
- Goldman, S.A.; Kuypers, N.J. How to make an oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Toyama, Y.; Nishiyama, A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002, 69, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Pickard, J.D.; Harris, N.G. Time course of cellular pathology after controlled cortical impact injury. Exp. Neurol. 2003, 182, 87–102. [Google Scholar] [CrossRef]
- Nascimento, D.; Pozza, D.H.; Castro-Lopes, J.M.; Neto, F.L. Neuronal injury marker ATF-3 Is induced in primary afferent neurons of monoarthritic rats. Neurosignals 2011, 19, 210–221. [Google Scholar] [CrossRef]
- Wong, A.W.; Osborne, P.B.; Keast, J.R. Axonal injury induces ATF3 in specific populations of sacral preganglionic neurons in male rats. Front. Neurosci. 2018, 12, 1–12. [Google Scholar] [CrossRef]
- Förstner, P.; Rehman, R.; Anastasiadou, S.; Haffner-Luntzer, M.; Sinske, D.; Ignatius, A.; Roselli, F.; Knöll, B. Neuroinflammation after traumatic brain injury is enhanced in activating transcription factor 3 mutant mice. J. Neurotrauma 2018, 35, 2317–2329. [Google Scholar] [CrossRef]
- Rostami, E.; Davidsson, J.; Chye Ng, K.; Lu, J.; Gyorgy, A.; Walker, J.; Wingo, D.; Plantman, S.; Bellander, B.M.; Agoston, D.V.; et al. A model for mild traumatic brain injury that induces limited transient memory impairment and increased levels of axon related serum biomarkers. Front. Neurol. 2012, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | |||
| Subjects | Trauma Level | Survival Time | Weight ± SD (g) |
| 18 Sprague-Dawley rats | Sham → n = 9 | 24 h → n = 3 | 411.7 ± 17.9 |
| 72 h → n = 3 | 429.7 ± 12.7 | ||
| 7 d → n = 3 | 399.7 ± 18.6 | ||
| 0.96–1.34 Mrad/s2 → n = 7 | 24 h → n = 3 | 444.7 ± 16.2 | |
| 72 h → n = 3 | 478 ± 88.7 | ||
| 7 d → n = 1 | 376 | ||
| 1.35–2.18 Mrad/s2 → n = 2 | 7 d → n = 2 | 393.5 ± 23.3 | |
| B | |||
| Subjects | Trauma Level | Survival Time | Weight ± SD (g) |
| 9 Sprague-Dawley rats | Sham → n = 2 | 24h → n = 1 | 372 |
| 72h → n = 1 | 354 | ||
| 0.96–1.34 Mrad/s2 → n = 1 | 72h → n = 1 | 749 | |
| 1.35–2.18 Mrad/s2 → n = 6 | 24h → n = 3 | 364.7 ± 9.0 | |
| 72h → n = 3 | 559.3 ± 181.4 | ||
| Antibody | Dilution | Supplier, Catalogue Number |
|---|---|---|
| Amyloid precursor protein (APP) | 1:200 | Invitrogen, 51-2700 |
| Myelin Basic Protein (MBP) | 1:200 | Merk Millipore, Ne1019 |
| Rip | 1:300 | DSHB |
| Olig2 | 1:500 | AbCam, Ab109186 |
| NG2 | 1:200 | Millipore, A25320 |
| Occludin | 1:100 | Santa Cruz, Sc-8145 |
| A2B5 (4D4 A2B2-like-supernatant) | 1:50 | DSHB |
| HSP70 | 1:200 | Santa Cruz, Sc-66048 |
| ATF3 | 1:100 | Santa Cruz, Sc-81189 |
| Target mRNA Probe | Nucleotides | Accession Number |
|---|---|---|
| Olig2 | 375-1986 | NM_001100557.1 |
| PDGFRa | 1516-2531 | NM_012802.1 |
| Survival Time | ROI | APP | MBP | Rip | OLIG2 | A2B5 | NG2 | PDGFRa | HSP70 | ATF3 |
|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | CC “SVZ” | ↑ | + | ↑ | + | + | + | + | - | - |
| CC “BORDER” | ↑ | + | ↑ | + | - | + | + | ↑ * | - | |
| CC “OCCIPITAL” | ↑ | + | + | ↓ | - | + | ↑ | ↑ * | - | |
| DORSAL CORTEX | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ | |
| HABENULAR NUCLEI | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ | |
| 72 h | CC “SVZ” | ↑ | + | + | + | + | + | + | - | - |
| CC “BORDER” | ↑ | + | + | ↓ | - | + | ↓ | - | - | |
| CC “OCCIPITAL” | ↑ | + | + | ↓ | + | ↑ | - | - | ||
| DORSAL CORTEX | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ | |
| HABENULAR NUCLEI | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ | |
| 7 d | CC “SVZ” | ↑ | + | ↑ | + | + | + | ↑ | - | - |
| CC “BORDER” | ↑ | + | + | + | - | + | ↓ | - | - | |
| CC “OCCIPITAL” | ↑ | + | + | ↓ | - | + | ↑ | - | - | |
| DORSAL CORTEX | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ | |
| HABENULAR NUCLEI | ↑ | N.I. | N.I. | N.I. | N.I. | N.I. | N.I. | ↑ | ↑ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losurdo, M.; Davidsson, J.; Sköld, M.K. Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury. Brain Sci. 2020, 10, 229. https://doi.org/10.3390/brainsci10040229
Losurdo M, Davidsson J, Sköld MK. Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury. Brain Sciences. 2020; 10(4):229. https://doi.org/10.3390/brainsci10040229
Chicago/Turabian StyleLosurdo, Michela, Johan Davidsson, and Mattias K. Sköld. 2020. "Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury" Brain Sciences 10, no. 4: 229. https://doi.org/10.3390/brainsci10040229
APA StyleLosurdo, M., Davidsson, J., & Sköld, M. K. (2020). Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury. Brain Sciences, 10(4), 229. https://doi.org/10.3390/brainsci10040229