Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Multiple Sclerosis (MS)
2.1. Immunopathophysiology of MS
2.2. Current Therapeutics of MS
2.3. Experimental Autoimmune Encephalomyelitis (EAE) as an Animal Model for MS
3. Nanotechnology and MS
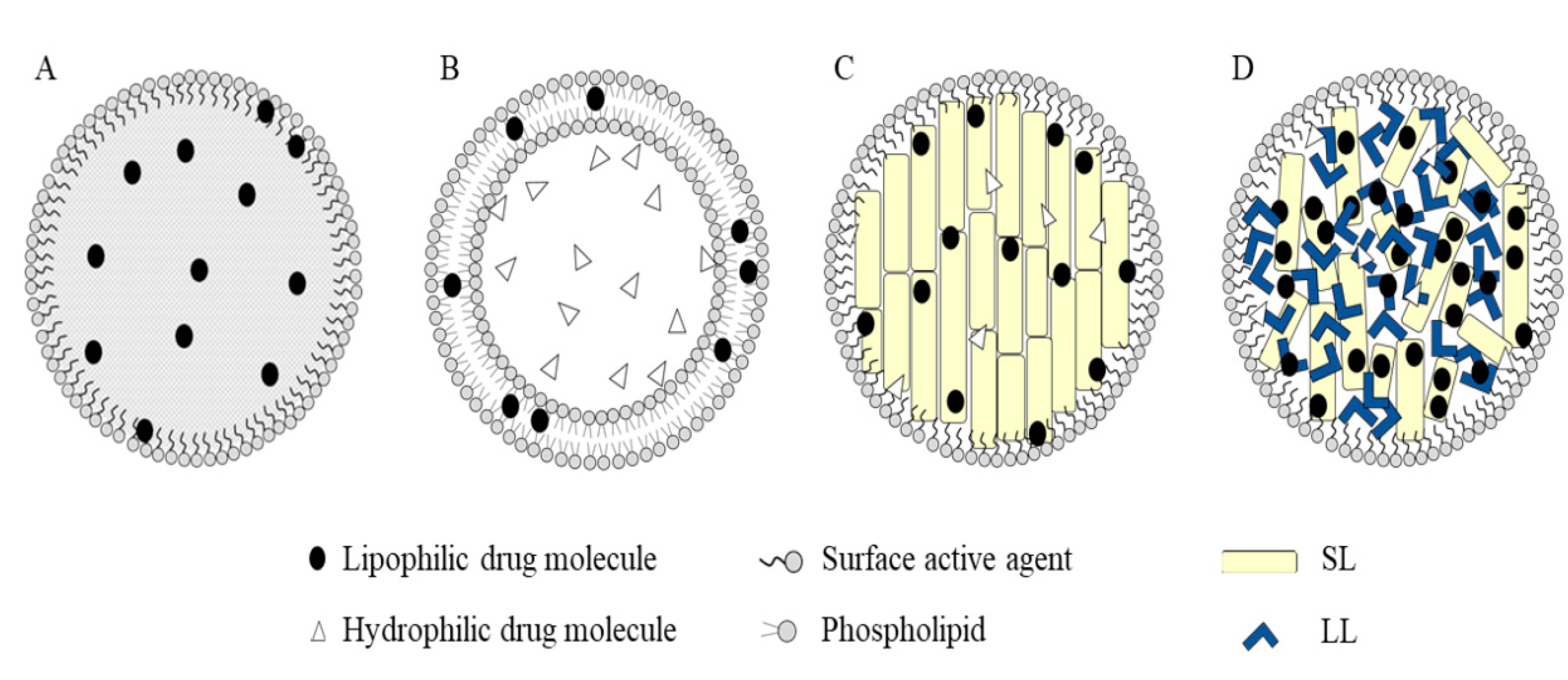
3.1. Lipid-Based Nanosystems
3.1.1. Nanolipid Carriers (NLCs) and Solid Lipid Nanoparticles (SLNs)
3.1.2. Liposomes
3.1.3. Other Lipid-Based Nanocarriers
3.2. Polymer-Based Nanosystems
3.2.1. Poly(Lactic-co-Glycolic Acid) (PLGA) Polymeric Nanoparticles
3.2.2. Other Polymeric Nanoparticles
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Passos, G.R.D.; Sato, D.K.; Becker, J.; Fujihara, K. Th17Cells pathways in multiple sclerosis and neuromyelitis optica spectrum disorders: Pathophysiological and therapeutic implications. Mediat. Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 2013, 9, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tillery, E.E.; Clements, J.N.; Howard, Z. What’s new in multiple sclerosis? Ment. Health Clin. 2018, 7, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Boissy, A.R.; Cohen, J.A. Multiple sclerosis symptom management. Expert Rev. Neurother. 2007, 7, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vecsei, L.; Milanov, I. The Epidemiology of Multiple Sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Diebold, M. Immunological treatment of multiple sclerosis. Semin Hematol. 2016, 53, 54–57. [Google Scholar] [CrossRef]
- Melchor, G.S.; Khan, T.; Reger, J.F.; Huang, J.K. Remyelination Pharmacotherapy Investigations Highlight Diverse Mechanisms Underlying Multiple Sclerosis Progression. ACS Pharmacol. Transl. Sci. 2019, 2, 372–386. [Google Scholar] [CrossRef]
- Ghalamfarsa, G.; Hojjat-Farsangi, M.; Mohammadnia-Afrouzi, M.; Anvari, E.; Farhadi, S.; Yousefi, M.; Jadidi-Niaragh, F. Application of nanomedicine for crossing the blood–brain barrier: Theranostic opportunities in multiple sclerosis. J. Immunotoxicol. 2016, 13, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Ballerini, C.; Baldi, G.; Aldinucci, A.; Maggi, P. Nanomaterial applications in multiple sclerosis inflamed brain. J. Neuroimmune Pharmacol. 2015, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gironi, M.; Arno, C.; Comi, G.; Penton-Rol, G.; Furlan, R. Chapter 4—Multiple Sclerosis and Neurodegenerative Diseases. In Immune Rebalancing: The Future of Immunosuppression; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 63–84. [Google Scholar]
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of multiple sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of Immune-Related Loci Identifies 48 New Susceptibility Variants for Multiple Sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.A.; Tzoulaki, I. Environmental Risk Factors and Multiple Sclerosis: An Umbrella Review of Systematic Reviews and Meta-Analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Hedström, A.K.; Åkerstedt, T.; Hillert, J.; Olsson, T.; Alfredsson, L. Shift Work at Young Age Is Associated with Increased Risk for Multiple Sclerosis. Ann. Neurol. 2011, 70, 733–741. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Ebers, G.C.; Dyment, D.A.; Risch, N.J. Evidence for genetic basis of multiple sclerosis. Lancet 1996, 347, 1728–1730. [Google Scholar] [CrossRef]
- Hemmer, B.; Nessler, S.; Zhou, D.; Kieseier, B.; Hartung, H.P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 2, 201–211. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Eckstein, C.; Bhatti, M.T. Currently approved and emerging oral therapies in multiple sclerosis: An update for the ophthalmologist. Surv. Ophthalmol. 2016, 61, 318–332. [Google Scholar] [CrossRef]
- Ian Douglas, J.K.C.; Rompani, P.; Singhal, B.S.; Thompson, A. Multiple sclerosis. In Neurological Disorders: Public Health Challenges; Saraceno, B., Ed.; WHO (World Health Organization): Geneva, Switzerland, 2005; pp. 85–94. [Google Scholar]
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain J. Neurol. 2017, 140, 527–546. [Google Scholar] [CrossRef] [Green Version]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; de Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple sclerosis: Immunopathology and treatment update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kallaur, A.P.; Lopes, J.; Oliveira, S.R.; Simão, A.N.; Reiche, E.M.; de Almeida, E.R.; Morimoto, H.K.; de Pereira, W.L. Immune-Inflammatory and oxidative and nitrosative stress biomarkers of depression symptoms in subjects with multiple sclerosis: Increased peripheral inflammation but less acute neuroinflammation. Mol. Neurobiol. 2015, 53, 5191–5202. [Google Scholar] [CrossRef] [PubMed]
- McFarland, H.F.; Martin, R. Multiple Sclerosis: A Complicated Picture of Autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kaskow, B.J.; Baecher-Allan, C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, 029025. [Google Scholar] [CrossRef]
- Farjam, M.; Zhang, G.X.; Ciric, B.; Rostami, A. Emerging immunopharmacological targets in multiple sclerosis. J. Neurol. Sci. 2015, 358, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.X.; Zhang, H.L.; Wu, X.J.; Zhu, J.; Ma, D.H.; Jin, T. Role of the immunogenic and tolerogenic subsets of dendritic cells in multiple sclerosis. Mediators Inflamm. 2015, 2015, 513295. [Google Scholar] [CrossRef]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Shegarfi, H.; Naddafi, F.; Mirshafiey, A. The role of natural killer cells in Alzheimer’s disease. Scand. J. Immunol. 2012, 76, 451–456. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Mirshafiey, A. Regulatory T-cell as orchestra leader in immunosuppression process of multiple sclerosis. Immunopharmacol. Immunotoxicol. 2011, 33, 545–567. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ghalamfarsa, G.; Hadinia, A.; Yousefi, M.; Jadidi-Niaragh, F. The role of natural killer T cells in B cell malignancies. Tumor Biol. 2013, 34, 1349–1360. [Google Scholar] [CrossRef]
- Mishra, M.K.; Yong, V.W. Myeloid Cells—Targets of Medication in Multiple Sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stys, P.K.; Tsutsui, S. Recent advances in understanding multiple sclerosis. F1000Res 2019, 8, F1000 Faculty Rev-2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingerchuk, D.M.; Carter, J.L. Multiple Sclerosis: Current and Emerging Disease-Modifying Therapies and Treatment Strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Cross, A.H.; Naismith, R.T. Established and Novel Disease-Modifying Treatments in Multiple Sclerosis. J. Intern. Med. 2014, 275, 350–363. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for Patients with Relapsing Multiple Sclerosis after Disease-Modifying Therapy: A Randomised Controlled Phase 3 Trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Katsara, M.; Matsoukas, J.; Deraos, G.; Apostolopoulos, V. Towards immunotherapeutic drugs and vaccines against multiple sclerosis. Acta Biochim. Biophys. Sin. 2008, 40, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E. Building different mouse models for human MS. Ann. N. Y. Acad. Sci. 2007, 1103, 11–18. [Google Scholar] [CrossRef]
- Burt, R.K.; Testori, A.; Craig, R.; Cohen, B.; Suffit, R.; Barr, W. Hematopoietic stem cell transplantation for autoimmune diseases: What have we learned? J. Autoimmun. 2008, 30, 116–120. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple Sclerosis: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, R.L.; Ifergan, I.; Miller, S.D. Experimental Autoimmune Encephalomyelitis in Mice. Methods Mol. Biol. 2016, 1304, 145–160. [Google Scholar] [PubMed] [Green Version]
- Sun, D.; Whitaker, J.N.; Huang, Z.; Liu, D.; Coleclough, C.; Wekerle, H.; Raine, C.S. Myelin Antigen-Specific CD8+ T Cells Are Encephalitogenic and Produce Severe Disease in C57BL/6 Mice. J. Immunol. 2001, 166, 7579–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal Models of Multiple Sclerosis: Focus on Experimental Autoimmune Encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The Experimental Autoimmune Encephalomyelitis (EAE) Model of MS. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 122, pp. 173–189. [Google Scholar]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple Sclerosis Animal Models: A Clinical and Histopathological Perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]
- Demetzos, C.; Pippa, N. Advanced drug delivery nanosystems (aDDnSs): A mini-review. Drug Deliv. 2014, 21, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.Á.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Shakya, A.K.; Nandakumar, K.S. Antigen-Specific Tolerization and Targeted Delivery as Therapeutic Strategies for Autoimmune Diseases. Trends Biotechnol. 2018, 36, 686–699. [Google Scholar] [CrossRef]
- Prinz, J.; Karacivi, A.; Stormanns, E.R.; Recks, M.S.; Kuerten, S. Time-Dependent Progression of Demyelination and Axonal Pathology in MP4-Induced Experimental Autoimmune Encephalomyelitis. PLoS ONE 2015, 10, e0144847. [Google Scholar] [CrossRef] [PubMed]
- Keough, M.B.; Yong, V.W. Remyelination therapy for multiple sclerosis. Neurotherapeutics 2013, 10, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni, R.; Herschkovitz, A.; Eilam, R.; Blumberg-Hazan, M.; Sela, M.; Bruck, W.; Arnon, R. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 11358–11363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Nakagomi, N.; Nakano-Doi, A.; Ishikawa, H.; Tatsumi, Y.; Bando, Y.; Yoshikawa, H.; Matsuyama, T.; Gomi, F.; Nakagomi, T. Potential of Adult Endogenous Neural Stem/Progenitor Cells in the Spinal Cord to Contribute to Remyelination in Experimental Autoimmune Encephalomyelitis. Cells 2019, 8, 1025. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, L.; Covacu, R.; Estrada, C.P.; Sankavaram, S.R.; Svensson, M.; Brundin, L. Long-distance effects of inflammation on differentiation of adult spinal cord neural stem/progenitor cells. J. Neuroimmunol. 2015, 288, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Münzel, E.J.; Williams, A. Promoting remyelination in multiple sclerosis—Recent advances. Drugs 2013, 73, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.P.; Zhang, J.Z.; Titus, H.E.; Karl, M.; Merzliakov, M.; Dorfman, A.R.; Karlik, S.; Stewart, M.G.; Watt, R.K.; Facer, B.D.; et al. Nanocatalytic activity of cleansurfaced, faceted nanocrystalline gold enhances remyelination in animal models of multiple sclerosis. Sci. Rep. 2020, 10, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.T.; Li, W.; Meng, G.; Wang, P.; Liao, W. Strategies for transporting nanoparticles across the blood-brain barrier. Biomater. Sci. 2016, 4, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Chen, J.; Gao, J. Nanocarriers as a powerful vehicle to overcome bloodbrain barrier in treating neurodegenerative diseases: Focus on recent advances. Asian J. Pharm. Sci. 2019, 14, 480–496. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [Green Version]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, M.; Saraf, S.; Saraf, S.; Dubey, S.K.; Puri, A.; Patel, R.J.; Ajazuddin; Ravichandiran, V.; Murty, U.S.; Amit, A. Recent strategies and advances in the fabrication of nano lipid carriers and their application towards brain targeting. J. Control. Release 2020, 321, 372–415. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Eldridge, D.; Palombo, E.; Harding, I. Composition and structure. In Lipid Nanoparticles: Production, Characterization and Stability; Shah, R., Eldridge, D., Palombo, E., Harding, I., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 11–22. [Google Scholar]
- Ganesan, P.; Narayanasamy, D. Lipid nanoparticles: Different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain. Chem. Pharm. 2017, 6, 37–56. [Google Scholar] [CrossRef]
- Gadhave, D.G.; Kokare, C.R. Nanostructured lipid carriers engineered for intranasal delivery of teriflunomide in multiple sclerosis: Optimization and in vivo studies. Drug Dev. Ind. Pharm. 2019, 45, 839–851. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, G.; Kumar, R.; Malik, R.; Singh, B.; Katare, O.P.; Raza, K. Enhanced brain delivery of dimethyl fumarate employing tocopherol-acetate-based nanolipidic carriers: Evidence from pharmacokinetic, biodistribution, and cellular uptake studies. ACS Chem. Neurosci. 2017, 8, 860–865. [Google Scholar] [CrossRef]
- Ghasemian, E.; Vatanara, A.; Navidi, N.; Rouini, M.R. Brain delivery of baclofen as a hydrophilic drug by nanolipid carriers: Characteristics and pharmacokinetics evaluation. J. Drug Deliv. Sci. Technol. 2017, 37, 67–73. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, G.; Gupta, V.; Kaur, R.; Thakur, K.; Malik, R.; Kumar, A.; Kaushal, N.; Katare, O.P.; Raza, K. Oral Delivery of Methylthioadenosine to the Brain Employing Solid Lipid Nanoparticles: Pharmacokinetic, Behavioral, and Histopathological Evidences. AAPS PharmSciTech. 2019, 20, 74. [Google Scholar] [CrossRef]
- Gandomi, N.; Varshochian, R.; Atyabi, F.; Ghahremani, M.H.; Sharifzadeh, M.; Amini, M.; Dinarvand, R. Solid lipid nanoparticles surface modified with anti-Contactin-2 or anti-Neurofascin for brain-targeted delivery of medicines. Pharm. Dev. Technol. 2017, 22, 426–435. [Google Scholar] [CrossRef]
- Chountoulesi, M.; Naziris, N.; Pippa, N.; Pispas, S.; Demetzos, C. Differential scanning calorimetry (DSC): An invaluable tool for the thermal evaluation of advanced chimeric liposomal drug delivery nanosystems. In Thermodynamics and Biophysics of Biomedical Nanosystems. Applications and Practical Considerations. Series: Series in BioEngineering; Demetzos, C., Pippa, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 297–337. [Google Scholar]
- Schmidt, J.; Metselaar, J.M.; Wauben, M.H.M.; Toyka, K.V.; Storm, G.; Gold, R. Drug targeting by long-circulating liposomal glucocorticosteroids increases therapeutic efficacy in a model of multiple sclerosis. Brain 2003, 126, 1895–1904. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, P.J.; Appeldoorn, C.C.; Rip, J.; Dorland, R.; van der Pol, S.M.; Kooij, G.; de Vries, H.E.; Reijerkerk, A. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J. Control. Release 2012, 164, 364–369. [Google Scholar] [CrossRef]
- Lee, D.H.; Rötger, C.; Appeldoorn, C.C.; Reijerkerk, A.; Gladdines, W.; Gaillard, P.J.; Linker, R.A. Glutathione PEGylated liposomal methylprednisolone (2B3-201) attenuates CNS inflammation and degeneration in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 96–101. [Google Scholar] [CrossRef]
- Turjeman, K.; Bavli, Y.; Kizelsztein, P.; Schilt, Y.; Allon, N.; Katzir, T.B.; Sasson, E.; Raviv, U.; Ovadia, H.; Barenholz, Y. Nano-Drugs Based on Nano Sterically Stabilized Liposomes for the Treatment of Inflammatory Neurodegenerative Diseases. PLoS ONE 2015, 10, e0130442. [Google Scholar] [CrossRef] [Green Version]
- Pujol-Autonell, I.; Mansilla, M.J.; Rodriguez-Fernandez, S.; Cano-Sarabia, M.; Navarro-Barriuso, J.; Ampudia, R.M.; Rius, A.; Garcia-Jimeno, S.; Perna-Barrull, D.; Martinez-Caceres, E.; et al. Liposome-based immunotherapy against autoimmune diseases: Therapeutic effect on multiple sclerosis. Nanomedicine 2017, 12, 1231–1242. [Google Scholar] [CrossRef]
- Binyamin, O.; Larush, L.; Frid, K.; Keller, G.; Friedman-Levi, Y.; Ovadia, H.; Abramsky, O.; Magdassi, S.; Gabizon, R. Treatment of a multiple sclerosis animal model by a novel nanodrop formulation of a natural antioxidant. Int. J. Nanomed. 2015, 10, 7165–7174. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Qi, S.; Chen, Y.; Luo, H.; Huang, S.; Yu, X.; Luo, Q.; Zhang, Z. Targeted immunomodulation of inflammatory monocytes across the blood-brain barrier by curcumin-loaded nanoparticles delays the progression of experimental autoimmune encephalomyelitis. Biomaterials 2020, 245, 119987. [Google Scholar] [CrossRef]
- Osorio-Querejeta, I.; Carregal-Romero, S.; Ayerdi-Izquierdo, A.; Mäger, I.; Nash, L.A.; Wood, M.; Egimendia, A.; Betanzos, M.; Alberro, A.; Iparraguirre, L.; et al. MiR-219a-5p Enriched Extracellular Vesicles Induce OPC Differentiation and EAE Improvement More Efficiently Than Liposomes and Polymeric Nanoparticles. Pharmaceutics 2020, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- El-Say, K.M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef]
- Houchin, M.L.; Topp, E.M. Chemical degradation of peptides and proteins in PLGA:a review of reactions and mechanisms. J. Pharm. Sci. 2008, 97, 2395–2404. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef]
- Samavedi, S.; Poindexter, L.K.; Van Dyke, M.; Goldstein, A.S. Synthetic biomaterials for regenerative medicine applications. Regen. Med. Appl. Organ. Transpl. 2014, 81–99. [Google Scholar] [CrossRef]
- Houchin, M.L.; Topp, E.M. Physical properties of PLGA films during polymer degradation. J. Appl. Polym. Sci. 2009, 114, 2848–2854. [Google Scholar] [CrossRef]
- Johnson, D.; Hafler, D.A.; Fallis, R.J.; Lees, M.B.; Brady, R.O.; Quarles, R.H.; Weiner, H.L. Cell-mediated immunity to myelin-associated glycoprotein, proteolipid pro-tein, and myelin basic protein in multiple sclerosis. J. Neuroimmunol. 1986, 13, 99–108. [Google Scholar] [CrossRef]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef]
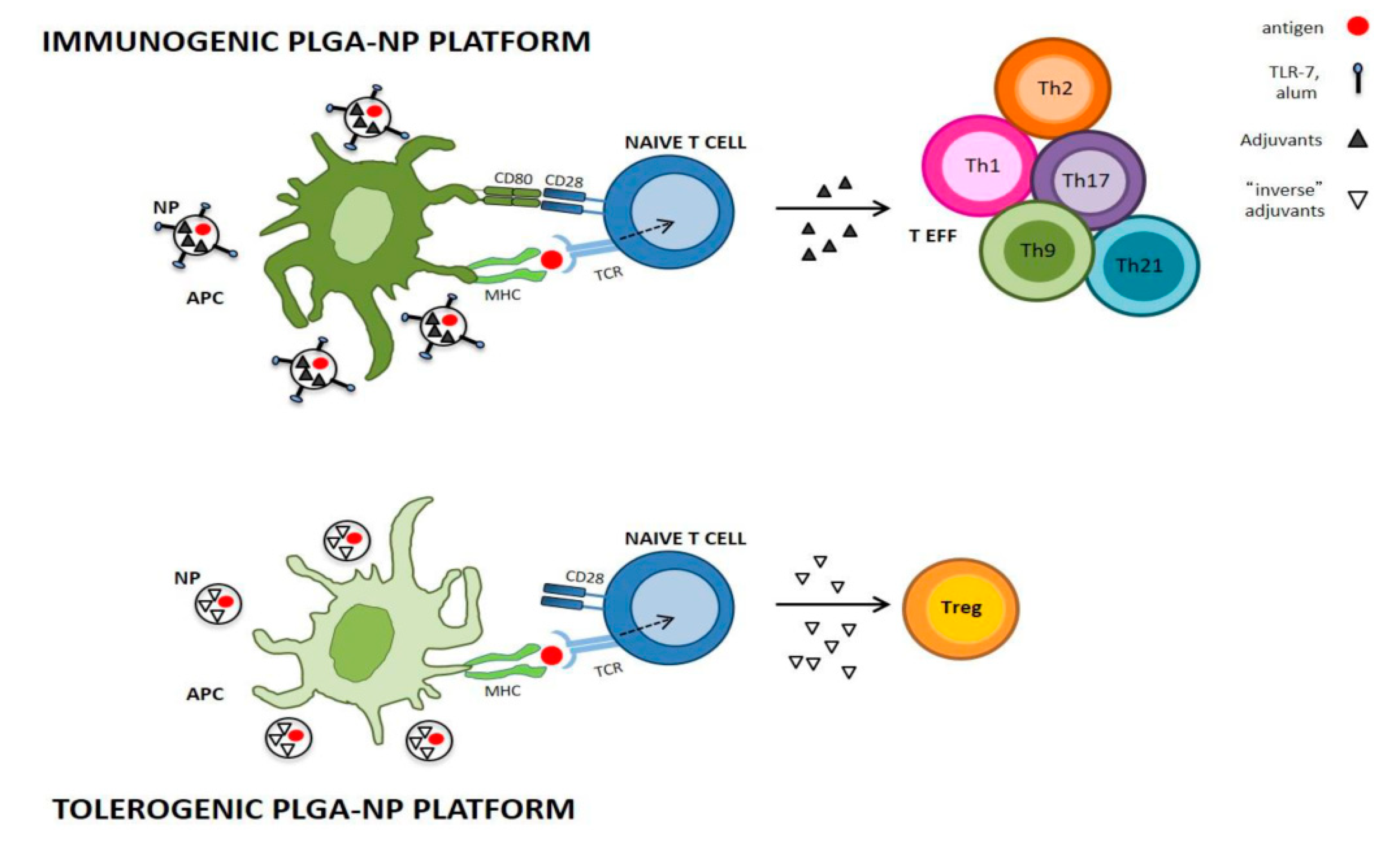
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, R.A.; LaMothe, R.A.; Ferrari, J.D.; Zhang, A.H.; Rossi, R.J.; Kolte, P.N.; Griset, A.P.; O’Neil, C.; Altreuter, D.H.; Browning, E.; et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.J.; Stewart, J.M.; Drashansky, T.T.; Brusko, M.A.; Zuniga, A.N.; Lorentsen, K.J.; Keselowsky, B.G.; Avram, D. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials 2017, 143, 79–92. [Google Scholar] [CrossRef]
- Pei, W.; Wan, X.; Shahzad, K.A.; Zhang, L.; Song, S.; Jin, X.; Wang, L.; Zhao, C.; Shen, C. Direct modulation of myelin-autoreactive CD4(+) and CD8(+) T cells in EAE mice by a tolerogenic nanoparticle co-carrying myelin peptide-loaded major histocompatibility complexes, CD47 and multiple regulatory molecules. Int. J. Nanomed. 2018, 13, 3731–3750. [Google Scholar] [CrossRef] [Green Version]
- Hunter, Z.; McCarthy, D.P.; Yap, W.T.; Harp, C.T.; Getts, D.R.; Shea, L.D.; Miller, S.D. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 2014, 8, 2148–2160. [Google Scholar] [CrossRef]
- Casey, L.M.; Pearson, R.M.; Hughes, K.R.; Liu, J.M.H.; Rose, J.A.; North, M.G.; Wang, L.Z.; Lei, M.; Miller, S.D.; Shea, L.D. Conjugation of Transforming Growth Factor Beta to Antigen-Loaded Poly(lactideco-glycolide) Nanoparticles Enhances Efficiency of Antigen-Specific Tolerance. Bioconjug. Chem. 2018, 29, 813–823. [Google Scholar] [CrossRef]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.; et al. Microparticles bearing encephalitogenic peptides induce T-cell toleranceand ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]
- Kuo, R.; Saito, E.; Miller, S.D.; Shea, L.D. Peptide-Conjugated Nanoparticles Reduce Positive Co-stimulatory Expression and T Cell Activity to Induce Tolerance. Mol. Ther. 2017, 25, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Saito, E.; Kuo, R.; Kramer, K.R.; Gohel, N.; Giles, D.A.; Moore, B.B.; Miller, S.D.; Shea, L.D. Design of biodegradable nanoparticles to modulate phenotypes of antigen-presenting cells for antigen-specific treatment of autoimmune disease. Biomaterials 2019, 222, 119432. [Google Scholar] [CrossRef]
- Rittchen, S.; Boyd, A.; Burns, A.; Park, J.; Fahmy, T.M.; Metcalfe, S.; Williams, A. Myelin repair in vivo is increased by targeting oligodendrocyte precursor cells with nanoparticles encapsulating leukaemia inhibitory factor (LIF). Biomaterials 2015, 56, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Führmann, T.; Ghosh, M.; Otero, A.; Goss, B.; Dargaville, T.R.; Pearse, D.D.; Dalton, P.D. Peptide-functionalized polymeric nanoparticles for active targeting of damaged tissue in animals with experimental autoimmune encephalomyelitis. Neurosci. Lett. 2015, 602, 126–132. [Google Scholar] [CrossRef]
- Al-Ghobashy, M.A.; El Meshad, A.N.; Abdelsalam, R.M.; Nooh, M.M.; Al-Shorbagy, M.; Laible, G. Development and Pre-Clinical Evaluation of Recombinant Human Myelin Basic Protein Nano Therapeutic Vaccine in Experimental Autoimmune Encephalomyelitis Mice Animal Model. Sci. Rep. 2017, 7, 46468. [Google Scholar] [CrossRef]
- Lunin, S.M.; Khrenov, M.O.; Glushkova, O.V.; Parfenyuk, S.B.; Novoselova, T.V.; Novoselova, E.G. Protective Effect of PBCA Nanoparticles Loaded with Thymulin Against the Relapsing-Remitting Form of Experimental Autoimmune Encephalomyelitis in Mice. Int. J. Mol. Sci. 2019, 20, 5374. [Google Scholar] [CrossRef] [Green Version]
- Kondiah, P.P.; Tomar, L.K.; Tyagi, C.; Choonara, Y.E.; Modi, G.; du Toit, L.C.; Kumar, P.; Pillay, V. A novel pH-sensitive interferon-β (INF-β) oral delivery system for application in multiple sclerosis. Int. J. Pharm. 2013, 456, 459–472. [Google Scholar] [CrossRef]
- Youssef, A.E.H.; Dief, A.E.; El Azhary, N.M.; Abdelmonsif, D.A.; El-fetiany, O.S. LINGO-1 siRNA nanoparticles promote central remyelination in ethidium bromide-induced demyelination in rats. J. Physiol. Biochem. 2019, 75, 89–99. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chountoulesi, M.; Demetzos, C. Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System. Brain Sci. 2020, 10, 338. https://doi.org/10.3390/brainsci10060338
Chountoulesi M, Demetzos C. Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System. Brain Sciences. 2020; 10(6):338. https://doi.org/10.3390/brainsci10060338
Chicago/Turabian StyleChountoulesi, Maria, and Costas Demetzos. 2020. "Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System" Brain Sciences 10, no. 6: 338. https://doi.org/10.3390/brainsci10060338
APA StyleChountoulesi, M., & Demetzos, C. (2020). Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System. Brain Sciences, 10(6), 338. https://doi.org/10.3390/brainsci10060338