Psychiatric Manifestation of Anti-LGI1 Encephalitis

 ,
,
Abstract
:1. Background
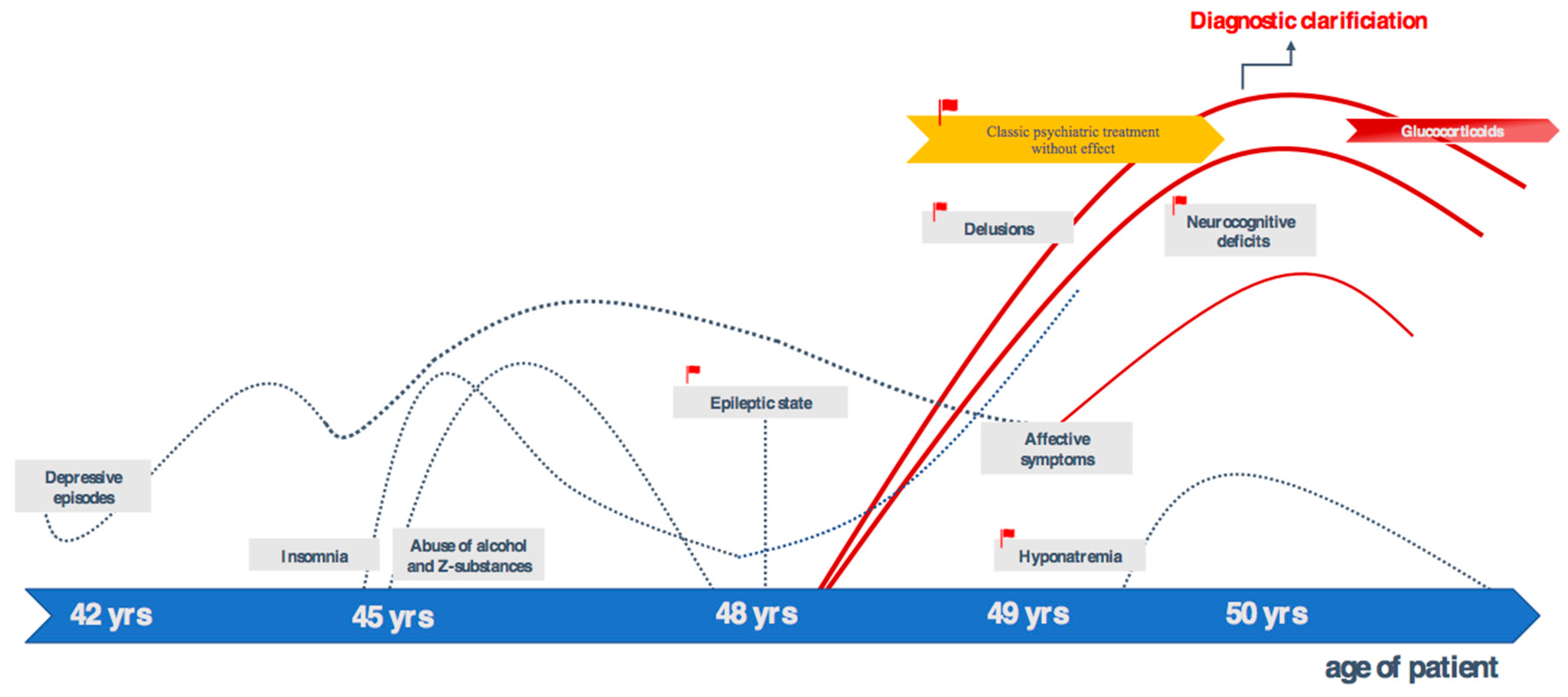
2. Case Presentation
3. Discussion
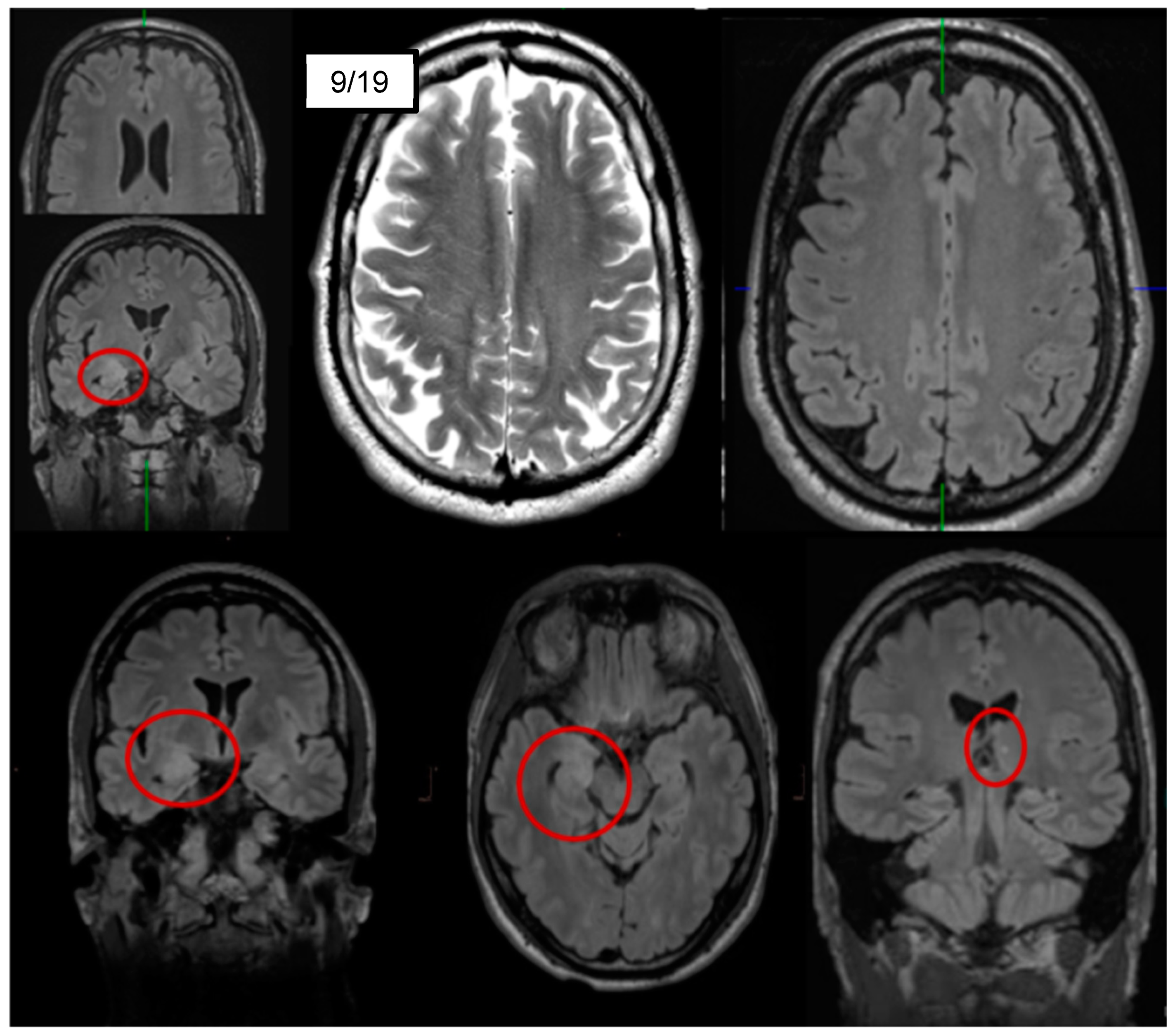
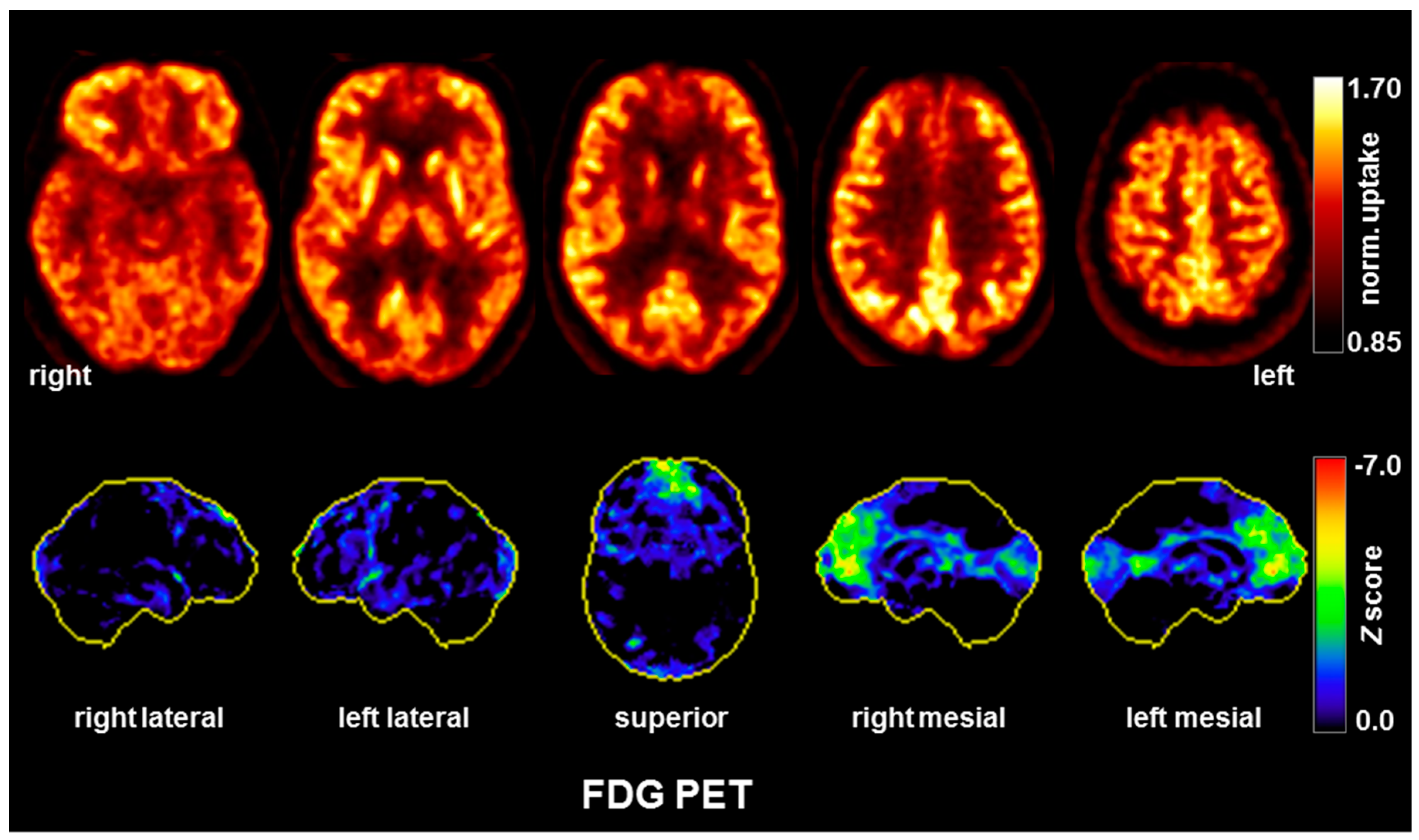
3.1. Diagnostic Assessment
3.2. Limitations
3.3. Clinical Consequences
3.4. Conceptual Considerations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.; Sillevis Smitt, P.A.; et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology 2016, 87, 1449–1456. [Google Scholar] [CrossRef]
- Van Sonderen, A.; Petit-Pedrol, M.; Dalmau, J.; Titulaer, M.J. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol. 2017, 13, 290–301. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F. Antibody-Mediated Encephalitis. N. Engl. J. Med. 2018, 378, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Van Sonderen, A.; Schreurs, M.W.; Wirtz, P.W.; Sillevis Smitt, P.A.; Titulaer, M.J. From VGKC to LGI1 and Caspr2 encephalitis: The evolution of a disease entity over time. Autoimmun Rev. 2016, 15, 970–974. [Google Scholar] [CrossRef] [Green Version]
- Prüss, H.; Lennox, B.R. Emerging psychiatric syndromes associated with antivoltage-gated potassium channel complex antibodies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef] [PubMed]
- Urbach, H.; Rauer, S.; Mader, I.; Paus, S.; Wagner, J.; Malter, M.P.; Prüss, H.; Lewerenz, J.; Kassubek, J.; Hegen, H.; et al. Supratentorial white matter blurring associated with voltage-gated potassium channel-complex limbic encephalitis. Neuroradiology 2015, 57, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, A.; Rauer, S.; Mader, I.; Meyer, P.T. Cerebral FDG-PET and MRI findings in autoimmune limbic encephalitis: Correlation with autoantibody types. J. Neurol. 2013, 260, 2744–2753. [Google Scholar] [CrossRef]
- Wegner, F.; Wilke, F.; Raab, P.; Tayeb, S.B.; Boeck, A.L.; Haense, C.; Trebst, C.; Voss, E.; Schrader, C.; Logemann, F.; et al. Anti-leucine rich glioma inactivated 1 protein and anti-N-methyl-D-aspartate receptor encephalitis show distinct patterns of brain glucose metabolism in 18F-fluoro-2-deoxy-d-glucose positron emission tomography. BMC Neurol. 2014, 14, 136. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Lee, S.T.; Bae, J.Y.; Kim, T.J.; Jun, J.S.; Moon, J.; Jung, K.H.; Park, K.I.; Irani, S.R.; Chu, K.; et al. LGI1 expression and human brain asymmetry: Insights from patients with LGI1-antibody encephalitis. J. Neuroinflamm. 2018, 15, 279. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Tripathi, M.; Roy, S.G.; Parida, G.K.; Ihtisham, K.; Dash, D.; Damle, N.; Shamim, S.A.; Bal, C. Metabolic topography of autoimmune non-paraneoplastic encephalitis. Neuroradiology 2018, 60, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Blinder, T.; Lewerenz, J. Cerebrospinal Fluid Findings in Patients with Autoimmune Encephalitis-A Systematic Analysis. Front Neurol. 2019, 10, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornau, H.C.; Kreye, J.; Stumpf, A.; Fukata, Y.; Parthier, D.; Sammons, R.P.; Imbrosci, B.; Kurpjuweit, S.; Kowski, A.B.; Fukata, M.; et al. Human Cerebrospinal Fluid Monoclonal LGI1 Autoantibodies Increase Neuronal Excitability. Ann. Neurol. 2020, 87, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Endres, D.; Maier, S.; Feige, B.; Mokhtar, N.B.; Nickel, K.; Goll, P.; Meyer, S.A.; Matthies, S.; Ebert, D.; Philipsen, A.; et al. Increased rates of intermittent rhythmic delta and theta activity in the electroencephalographies of adult patients with attention-deficit hyperactivity disorder. Epilepsy Behav. 2017, 75, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Salmon, E.; Garraux, G.; Delbeuck, X.; Collette, F.; Kalbe, E.; Zuendorf, G.; Perani, D.; Fazio, F.; Herholz, K. Predominant ventromedial frontopolar metabolic impairment in frontotemporal dementia. Neuroimage 2003, 20, 435–440. [Google Scholar] [CrossRef]
- Minoshima, S.; Frey, K.A.; Koeppe, R.A.; Foster, N.L.; Kuhl, D.E. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J. Nucl. Med. 1995, 36, 1238–1248. [Google Scholar]
- Heine, J.; Prüss, H.; Bartsch, T.; Ploner, C.J.; Paul, F.; Finke, C. Imaging of autoimmune encephalitis—Relevance for clinical practice and hippocampal function. Neuroscience 2015, 309, 68–83. [Google Scholar] [CrossRef]
- Dodich, A.; Cerami, C.; Iannaccone, S.; Marcone, A.; Alongi, P.; Crespi, C.; Canessa, N.; Andreetta, F.; Falini, A.; Cappa, S.F.; et al. Neuropsychological and FDG-PET profiles in VGKC autoimmune limbic encephalitis. Brain Cogn. 2016, 108, 81–87. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Zhang, C.; Cui, T.; Shi, W.X.; Guan, H.Z.; Ren, H.T.; Shao, X.Q. Seizure semiology in leucine-rich glioma-inactivated protein 1 antibody-associated limbic encephalitis. Epilepsy Behav. 2017, 77, 90–95. [Google Scholar] [CrossRef]
- Shan, W.; Liu, X.; Wang, Q. Teaching NeuroImages: 18F-FDG-PET/SPM analysis in 3 different stages from a patient with LGI-1 autoimmune encephalitis. Neurology 2019, 93, e1917–e1918. [Google Scholar] [CrossRef] [PubMed]
- Newey, C.R.; Sarwal, A. Hyponatremia and Voltage Gated Potassium Channel Antibody Associated Limbic Encephalitis. J. Neurol Neurophysiol. 2014, 5, 195. [Google Scholar] [CrossRef] [Green Version]
- Black, N.; Hamada, H. Possible anti-VGKC autoimmune limbic encephalitis associated with SIADH. BMJ Case Rep. 2018, bcr–2017. [Google Scholar] [CrossRef] [PubMed]
- Head, K.; Gong, S.; Joseph, S.; Wang, C.; Burkhardt, T.; Rossi, M.R.; LaDuca, J.; Matsui, S.; Vaughan, M.; Hicks, D.G.; et al. Defining the expression pattern of the LGI1 gene in BAC transgenic mice. Mamm. Genome. 2007, 18, 328–337. [Google Scholar] [CrossRef]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Prüß, H.; Köhler, S.; Müller, S. Autoimmune encephalitis-Diagnostic and therapeutic decision tree from a psychiatric, neurological and ethico-legal point of view: Approach in cases of lack of ability to give consent and permissibility of compulsory treatment. Der Nervenarzt 2020, 91, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.S.H.; Ho, R.C.M.; Quek, A.M.L. Chronic Manganese Toxicity Associated with Voltage-Gated Potassium Channel Complex Antibodies in a Relapsing Neuropsychiatric Disorder. Int. J. Environ. Res. Public Health 2018, 15, 783. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hao, Q.; He, L.; Wang, Q. LGI1 antibody encephalitis and psychosis. Australas. Psychiatry 2018, 26, 612–614. [Google Scholar] [CrossRef]
- Ellul, P.; Groc, L.; Tamouza, R.; Leboyer, M. The Clinical Challenge of Autoimmune Psychosis: Learning from Anti-NMDA Receptor Autoantibodies. Front Psychiatry 2017, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Herken, J.; Prüss, H. Red Flags: Clinical Signs for Identifying Autoimmune Encephalitis in Psychiatric Patients. Front Psychiatry 2017, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Najjar, S.; Steiner, J.; Najjar, A.; Bechter, K. A clinical approach to new-onset psychosis associated with immune dysregulation: The concept of autoimmune psychosis. J. Neuroinflamm. 2018, 15, 40. [Google Scholar] [CrossRef]
- Endres, D.; Bechter, K.; Prüss, H.; Hasan, A.; Steiner, J.; Leypoldt, F.; Tebartz van Elst, L. Autoantibody-associated schizophreniform psychoses: Clinical symptomatology. Der Nervenarzt 2019, 90, 547–563, German. [Google Scholar] [CrossRef]
- Tebartz van Elst, L.; Bechter, K.; Prüss, H.; Hasan, A.; Steiner, J.; Leypoldt, F.; Endres, D. Autoantibody-associated schizophreniform psychoses: Pathophysiology, diagnostics, and treatment. Der Nervenarzt 2019, 90, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, T.A.; Lennox, B.R.; Müller, S.; Benros, M.E.; Prüss, H.; Tebartz van Elst, L.; Klein, H.; Steiner, J.; Frodl, T.; Bogerts, B.; et al. Autoimmune psychosis: An international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry 2020, 7, 93–108. [Google Scholar] [CrossRef]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef] [Green Version]
- McKeon, A.; Marnane, M.; O’connell, M.; Stack, J.P.; Kelly, P.J.; Lynch, T. Potassium channel antibody associated encephalopathy presenting with a frontotemporal dementia like syndrome. Arch. Neurol. 2007, 64, 1528–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppiah, R.; Mukhtyar, C.; Flossmann, O.; Alberici, F.; Baslund, B.; Batra, R.; Brown, D.; Holle, J.; Hruskova, Z.; Jayne, D.R.; et al. A cross-sectional study of the Birmingham Vasculitis Activity Score version 3 in systemic vasculitis. Rheumatology (Oxf.) 2011, 50, 899–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, D.; Perlov, E.; Stich, O.; Rauer, S.; Maier, S.; Waldkircher, Z.; Lange, T.; Mader, I.; Meyer, P.T.; van Elst, L.T. Hypoglutamatergic state is associated with reduced cerebral glucose metabolism in anti-NMDA receptor encephalitis: A case report. BMC Psychiatry 2015, 15, 186. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Neuropsychiatric examination |
|
| Blood analyses |
|
| Cerebrospinal fluid analyses (was performed three times after in-patient admission) |
|
| Cerebral magnetic resonance imaging |
|
| Electroencephalo-graphy |
|
| FDG-PET |
|
| Heart examination |
|
| Computer tomography and x-ray thorax |
|
| Bronchoscopy |
|
| Sonography of the abdomen |
|
| Electromyography |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endres, D.; Prüss, H.; Dressing, A.; Schneider, J.; Feige, B.; Schweizer, T.; Venhoff, N.; Nickel, K.; Meixensberger, S.; Matysik, M.; et al. Psychiatric Manifestation of Anti-LGI1 Encephalitis. Brain Sci. 2020, 10, 375. https://doi.org/10.3390/brainsci10060375
Endres D, Prüss H, Dressing A, Schneider J, Feige B, Schweizer T, Venhoff N, Nickel K, Meixensberger S, Matysik M, et al. Psychiatric Manifestation of Anti-LGI1 Encephalitis. Brain Sciences. 2020; 10(6):375. https://doi.org/10.3390/brainsci10060375
Chicago/Turabian StyleEndres, Dominique, Harald Prüss, Andrea Dressing, Johanna Schneider, Bernd Feige, Tina Schweizer, Nils Venhoff, Kathrin Nickel, Sophie Meixensberger, Miriam Matysik, and et al. 2020. "Psychiatric Manifestation of Anti-LGI1 Encephalitis" Brain Sciences 10, no. 6: 375. https://doi.org/10.3390/brainsci10060375
APA StyleEndres, D., Prüss, H., Dressing, A., Schneider, J., Feige, B., Schweizer, T., Venhoff, N., Nickel, K., Meixensberger, S., Matysik, M., Maier, S. J., Domschke, K., Urbach, H., Meyer, P. T., & Tebartz van Elst, L. (2020). Psychiatric Manifestation of Anti-LGI1 Encephalitis. Brain Sciences, 10(6), 375. https://doi.org/10.3390/brainsci10060375