Parkinsonian Syndrome with Frontal Lobe Involvement and Anti-Glycine Receptor Antibodies

 ,
,
Abstract
:1. Background
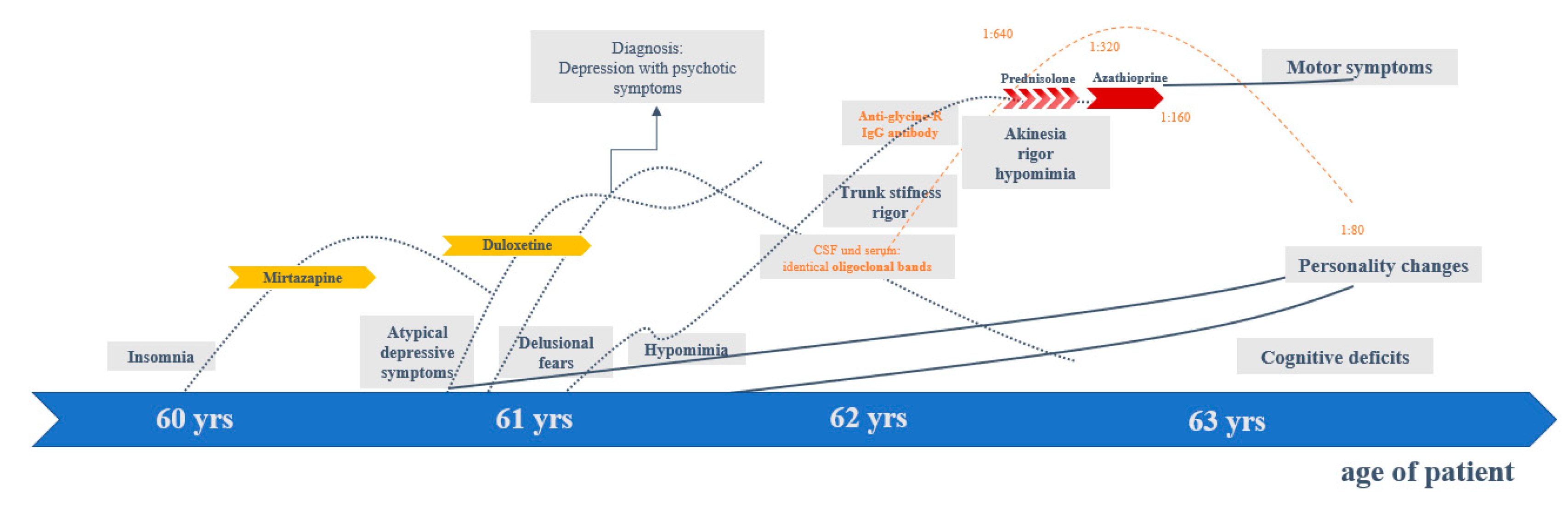
2. Case Presentation
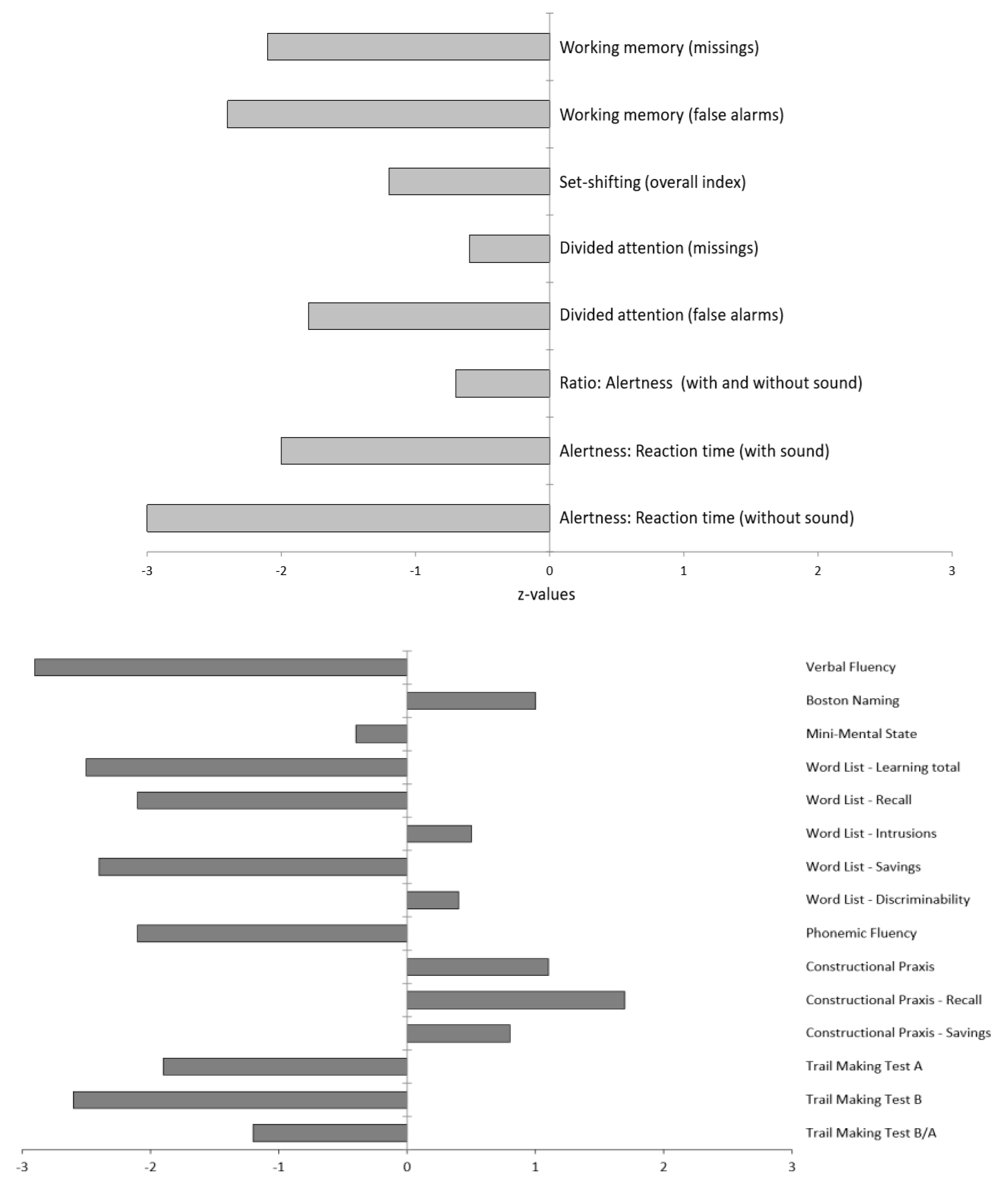
2.1. Diagnostic Findings
2.2. Illness, Somatic, and Family History
3. Discussion
- The anti-GlyR antibodies could have been generated first, thereby triggering a clinical phenotype as a Parkinsonian syndrome with frontal lobe involvement. This is conceivable in principle, but very unlikely in the presented patient; no typical signs of autoimmune anti-GlyR antibody syndrome presently known (e.g., hyperexcitability, anti-GlyR antibodies in CSF, or other inflammatory CSF changes) were detected [17]. Furthermore, the instrument-based diagnostics, using EEG, MRI and FDG PET, as well as the poor response to anti-inflammatory treatment did not support the hypothesis of underlying autoimmune encephalitis.
- The joint presence of a Parkinsonian syndrome with frontal lobe involvement and anti-GlyR antibodies could have been a pure coincidence. In this case, the anti-GlyR antibodies might be of no relevance. From the authors’ perspective, according to current knowledge, this alternative is possible. In antibody prevalence studies of healthy individuals, anti-GlyR antibodies have also been found in the serum of 0.06% of healthy individuals [one subject in a healthy control group of 1703 individuals [30].
- Finally, a Parkinsonian syndrome with secondary anti-GlyR antibody production is plausible. In this case, the anti-GlyR antibodies could even have effects on the brain, but these would likely be overlooked in the progressive neurodegenerative Parkinsonian syndrome. In the authors’ view, this seems a plausible scenario. Indeed, a similar discussion is currently taking place regarding anti-IgLON5 antibodies; in patients with increased levels of anti-IgLON5 antibodies, a tauopathy of the brainstem tegmentum has been described in combination with antibody-mediated encephalopathy that is responsive to immunotherapy [22,23]. According to these novel observations, a new basic principle of interaction between neurodegeneration and neuroimmunological processes is conceivable. In the present case, it would be similar to encephalopathy with IgLON5 antibodies; antineuronal antibodies possibly modulate the clinical course of PSP/CBD or other neurodegenerative disorders, without primarily causing those disorders.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grayson, M. Parkinson’s disease. Nature 2016, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxer, A.L.; Yu, J.; Golbe, I.L.; Litvan, I.; Lang, E.A.; Höglinger, G.U. Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017, 16, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Hufschmidt, A.; Lücking, C.; Rauer, S.; Glocker, F.X. Neurologie Compact; Hrsg. 7. Auflage.; Thieme: Stuttgart, Germany, 2017. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, E.A.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef]
- Meyer, P.; Hellwig, S. Update on SPECT and PET in parkinsonism—Part 1. Curr. Opin. Neurol. 2014, 27, 390–397. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Litvan, I.; Lang, E.A.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Parmera, J.B.; Rodriguez, R.D.; Neto, A.S.; Nitrini, R.; Brucki, S.M.D. Corticobasal syndrome: A diagnostic conundrum. Dement. Neuropsychol. 2016, 10, 267–275. [Google Scholar] [CrossRef]
- Mimuro, M.; Yoshida, M. Chameleons and mimics: Progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2019, 40, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, M.; Tang, C.C.; Feigin, A.; Allen, P.J.; Heinen, L.; Hellwig, S.; Amtage, F.; Hanspal, E.; Vonsattel, J.P.; Poston, K.L.; et al. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain 2014, 137, 3036–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, P.; Frings, L.; Rücker, G.; Hellwig, S. 18F-FDG PET in Parkinsonism: Differential Diagnosis and Evaluation of Cognitive Impairment. J. Nucl. Med. 2017, 58, 1888–1898. [Google Scholar] [CrossRef] [Green Version]
- Kannoth, S.; Anandakkuttan, A.; Mathai, A.; Sasikumar, A.N.; Nambiar, V.; Anandakuttan, A. Autoimmune atypical parkinsonism—A group of treatable parkinsonism. J. Neurol. Sci. 2016, 362, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Vincent, A.; Meinck, H.-M.; Irani, S.R.; Bhatia, K.P. Movement disorders with neuronal antibodies: Syndromic approach, genetic parallels and pathophysiology. Brain 2017, 141, 13–36. [Google Scholar] [CrossRef]
- Endres, D.; Bechter, K.; Prüss, H.; Hasan, A.; Steiner, J.; Leypoldt, F.; Tebartz van Elst, L. Autoantikörper-assoziierte schizophreniforme Psychosen: Klinische Symptomatik. Der Nervenarzt 2019, 90, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Endres, D.; Leypoldt, F.; Bechter, K.; Hasan, A.; Steiner, J.; Domschke, K.; Wandinger, K.-P.; Falkai, P.; Arolt, V.; Stich, O.; et al. Autoimmune encephalitis as a differential diagnosis of schizophreniform psychosis: Clinical symptomatology, pathophysiology, diagnostic approach, and therapeutic considerations. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, M.E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.-M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- Piquet, A.L.; Khan, M.; Warner, J.E.; Wicklund, M.P.; Bennett, J.L.; Leehey, M.A.; Seeberger, L.; Schreiner, T.L.; Soldan, M.M.P.; Clardy, S.L. Novel clinical features of glycine receptor antibody syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e592. [Google Scholar] [CrossRef] [Green Version]
- Blinder, T.; Lewerenz, J. Cerebrospinal Fluid Findings in Patients with Autoimmune Encephalitis-A Systematic Analysis. Front. Neurol. 2019, 10, 804. [Google Scholar] [CrossRef] [Green Version]
- McKeon, A.; Martinez-Hernandez, E.; Lancaster, E.; Matsumoto, J.Y.; Harvey, R.J.; McEvoy, K.M.; Pittock, S.J.; Lennon, V.A.; Dalmau, J. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013, 70, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Swayne, A.; Tjoa, L.; Broadley, S.; Dionisio, S.; Gillis, D.; Jacobson, L.; Woodhall, M.R.; McNabb, A.; Schweitzer, D.; Tsang, B.; et al. Antiglycine receptor antibody related disease: A case series and literature review. Eur. J. Neurol. 2018, 25, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoshima, S.; Frey, A.K.; A Koeppe, R.; Foster, N.L.; Kuhl, D.E. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J. Nucl. Med. 1995, 36, 1238–1248. [Google Scholar]
- Whitwell, J.L.; Jack, C.R.; Boeve, B.F.; Parisi, J.E.; Ahlskog, J.E.; Drubach, D.A.; Senjem, M.L.; Knopman, D.S.; Petersen, R.C.; Dickson, D.W.; et al. Imaging correlates of pathology in corticobasal syndrome. Neurology 2010, 75, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Massey, L.A.; Jäger, H.R.; Paviour, D.C.; O’Sullivan, S.S.; Ling, H.; Williams, D.R.; Kallis, C.; Holton, J.; Revesz, T.; Burn, D.; et al. The midbrain to pons ratio: A simple and specific MRI sign of progressive supranuclear palsy. Neurology 2013, 80, 1856–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouri, N.; Whitwell, J.L.; Josephs, K.A.; Rademakers, R.; Dickson, D.W. Corticobasal degeneration: A pathologically distinct 4R tauopathy. Nat. Rev. Neurol. 2011, 7, 263–272. [Google Scholar] [CrossRef]
- Hinson, S.R.; López-Chiriboga, A.S.; Bower, J.H.; Matsumoto, J.Y.; Hassan, A.; Basal, E.; Lennon, V.A.; Pittock, S.J.; McKeon, A. Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e438. [Google Scholar] [CrossRef] [Green Version]
- Crisp, S.J.; Dixon, C.L.; Jacobson, L.; Chabrol, E.; Irani, S.R.; Leite, M.I.; Leschziner, G.; Slaght, S.J.; Vincent, A.; Kullmann, D.M. Glycine receptor autoantibodies disrupt inhibitory neurotransmission. Brain 2019, 142, 3398–3410. [Google Scholar] [CrossRef]
- Dahm, L.; Ott, C.; Steiner, J.; Stepniak, B.; Teegen, B.; Saschenbrecker, S.; Hammer, C.; Borowski, K.; Begemann, M.; Lemke, S.; et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann. Neurol. 2014, 76, 82–94. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood analyses |
|
| Cerebrospinal fluid analyses |
|
| Cerebral magnetic resonance imaging |
|
| Spinal magnetic resonance imaging |
|
| Electroencephalo-graphy. |
|
| [18F]fluorodeoxy-glucose positron emission tomography (FDG PET) |
|
| [123]FPCIT single-photon emission computed tomography (FPCIT SPECT) |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endres, D.; Prüss, H.; Rijntjes, M.; Schweizer, T.; Werden, R.; Nickel, K.; Meixensberger, S.; Runge, K.; Urbach, H.; Domschke, K.; et al. Parkinsonian Syndrome with Frontal Lobe Involvement and Anti-Glycine Receptor Antibodies. Brain Sci. 2020, 10, 399. https://doi.org/10.3390/brainsci10060399
Endres D, Prüss H, Rijntjes M, Schweizer T, Werden R, Nickel K, Meixensberger S, Runge K, Urbach H, Domschke K, et al. Parkinsonian Syndrome with Frontal Lobe Involvement and Anti-Glycine Receptor Antibodies. Brain Sciences. 2020; 10(6):399. https://doi.org/10.3390/brainsci10060399
Chicago/Turabian StyleEndres, Dominique, Harald Prüss, Michel Rijntjes, Tina Schweizer, Rita Werden, Kathrin Nickel, Sophie Meixensberger, Kimon Runge, Horst Urbach, Katharina Domschke, and et al. 2020. "Parkinsonian Syndrome with Frontal Lobe Involvement and Anti-Glycine Receptor Antibodies" Brain Sciences 10, no. 6: 399. https://doi.org/10.3390/brainsci10060399
APA StyleEndres, D., Prüss, H., Rijntjes, M., Schweizer, T., Werden, R., Nickel, K., Meixensberger, S., Runge, K., Urbach, H., Domschke, K., Meyer, P. T., & Tebartz van Elst, L. (2020). Parkinsonian Syndrome with Frontal Lobe Involvement and Anti-Glycine Receptor Antibodies. Brain Sciences, 10(6), 399. https://doi.org/10.3390/brainsci10060399