Potential Effects of Poloxamer 188 on Rat Isolated Brain Mitochondria after Oxidative Stress In Vivo and In Vitro



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (a)
- Developing an in vitro I/R injury model through exposure of rat isolated forebrain mitochondria to the ROS hydrogen peroxide (H2O2) after isolation;
- (b)
- Developing an in vivo I/R injury model through asphyxial cardiac arrest (CA) in rats before isolation of forebrain mitochondria;
- (c)
- Assessing potential positive effects of P188 on isolated, injured mitochondria by measuring ATP synthesis, O2 consumption, and calcium retention capacity (CRC).
2. Materials and Methods
2.1. Animals
2.1.1. Animal Preparation for Euthanasia
2.1.2. Animal Preparation for Asphyxial CA
2.1.3. Brain Extraction
2.2. Isolation of Mitochondria
2.3. Determination of Mitochondrial Protein Concentration
2.4. Injuring Mitochondria
2.4.1. In Vitro Injury with H2O2
2.4.2. In Vivo Injury by Asphyxial CA
2.5. Treatment with P188
2.5.1. After H2O2-Induced Injury In Vitro
2.5.2. After Asphyxial CA-Induced Injury In Vivo
2.6. Assessment of Mitochondrial Function Parameters
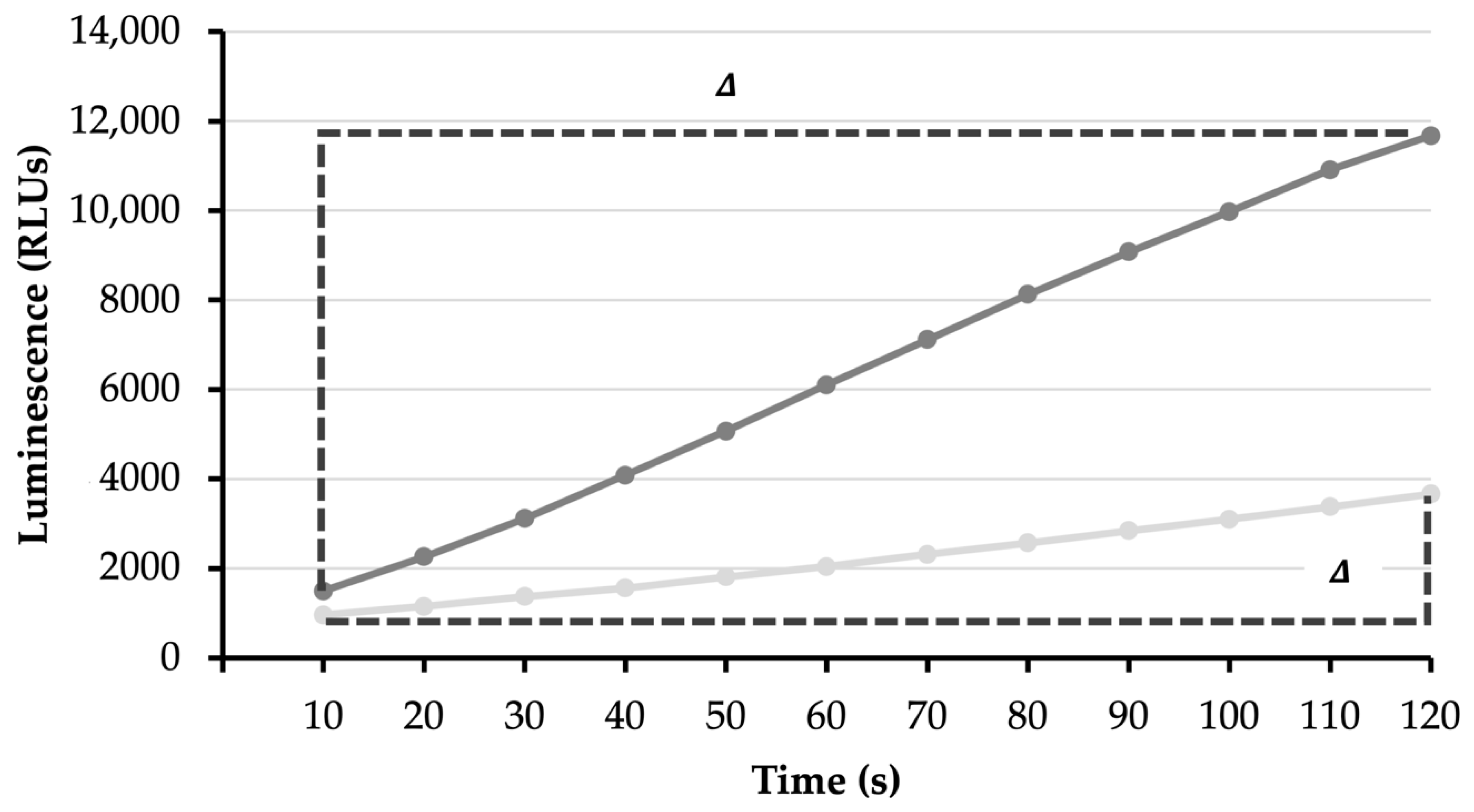
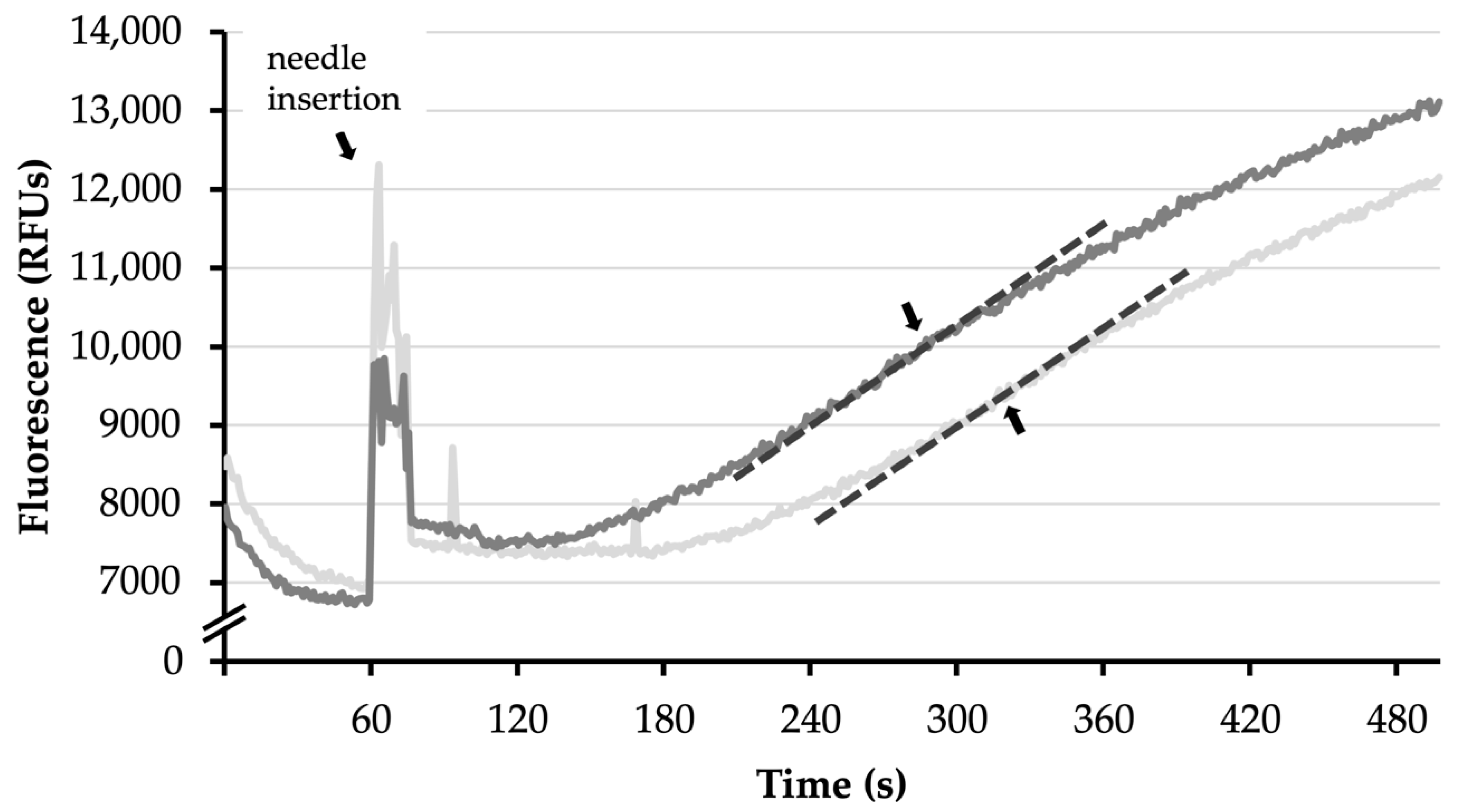
2.6.1. ATP Synthesis Assay
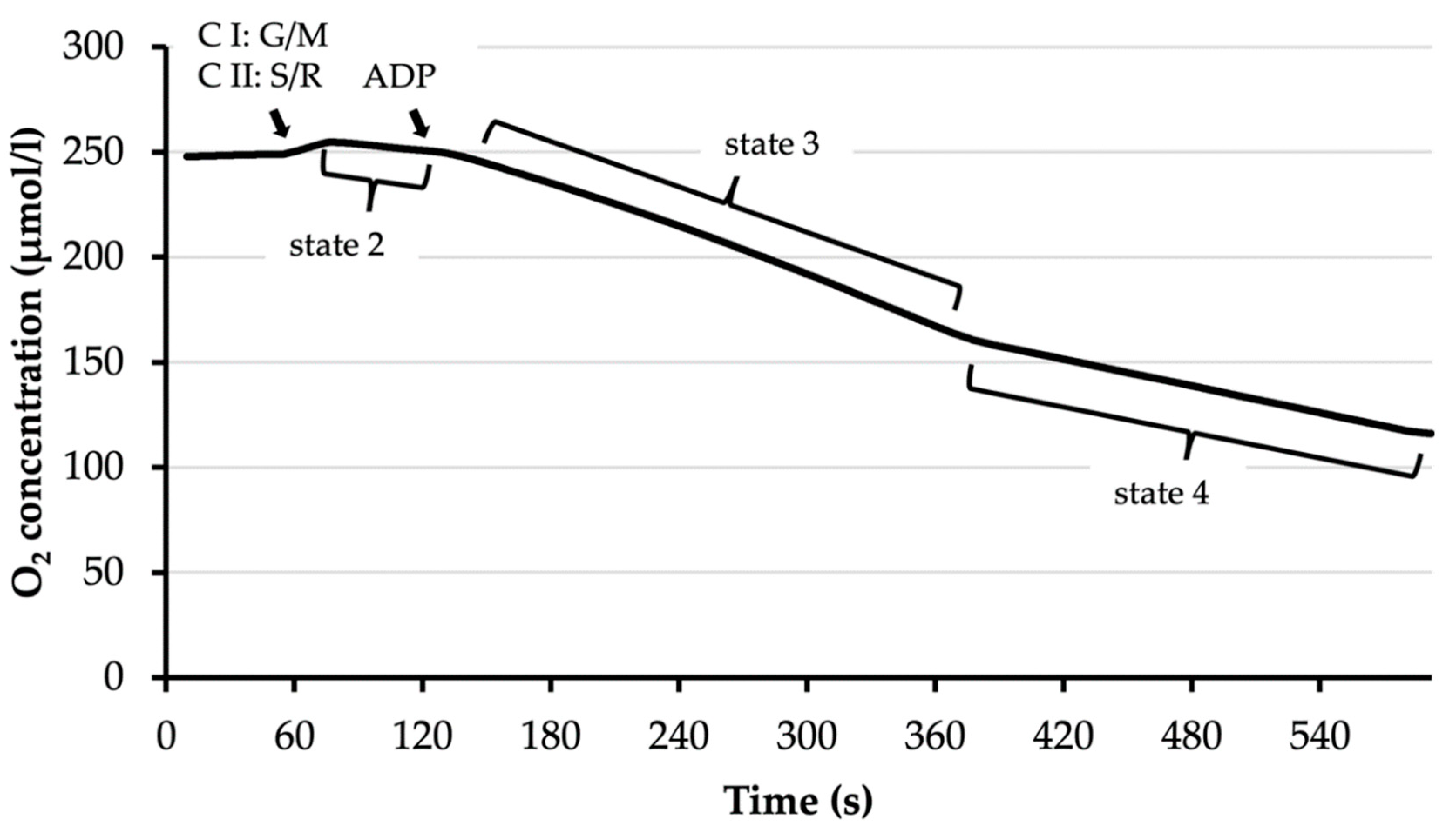
2.6.2. O2 Consumption Assay
2.6.3. CRC Assay
2.7. Data Analysis and Statistics
3. Results
3.1. Isolated Mitochondria after H2O2-Induced Injury
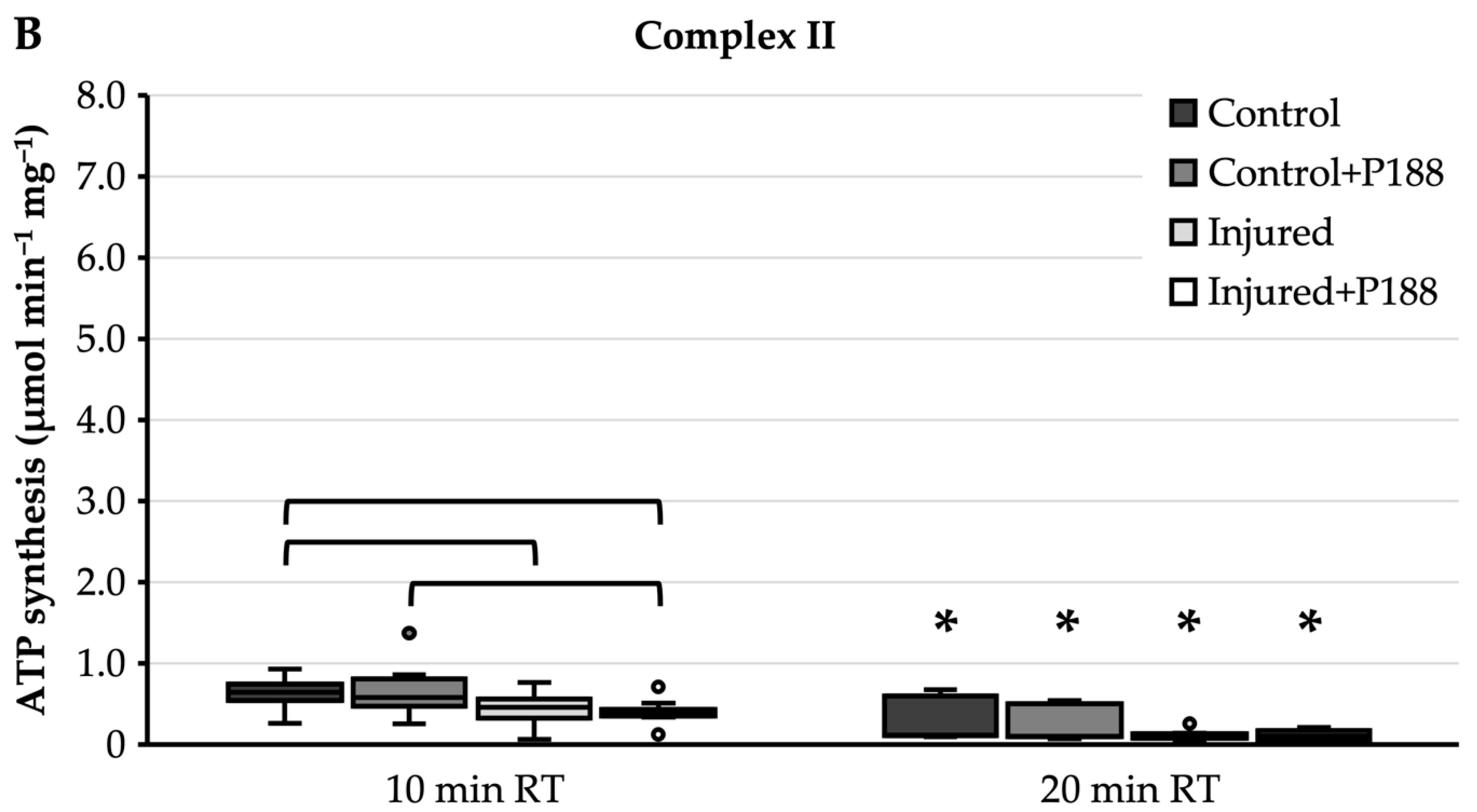
3.1.1. ATP Synthesis after H2O2-Induced Injury
3.1.2. O2 Consumption after H2O2-Induced Injury
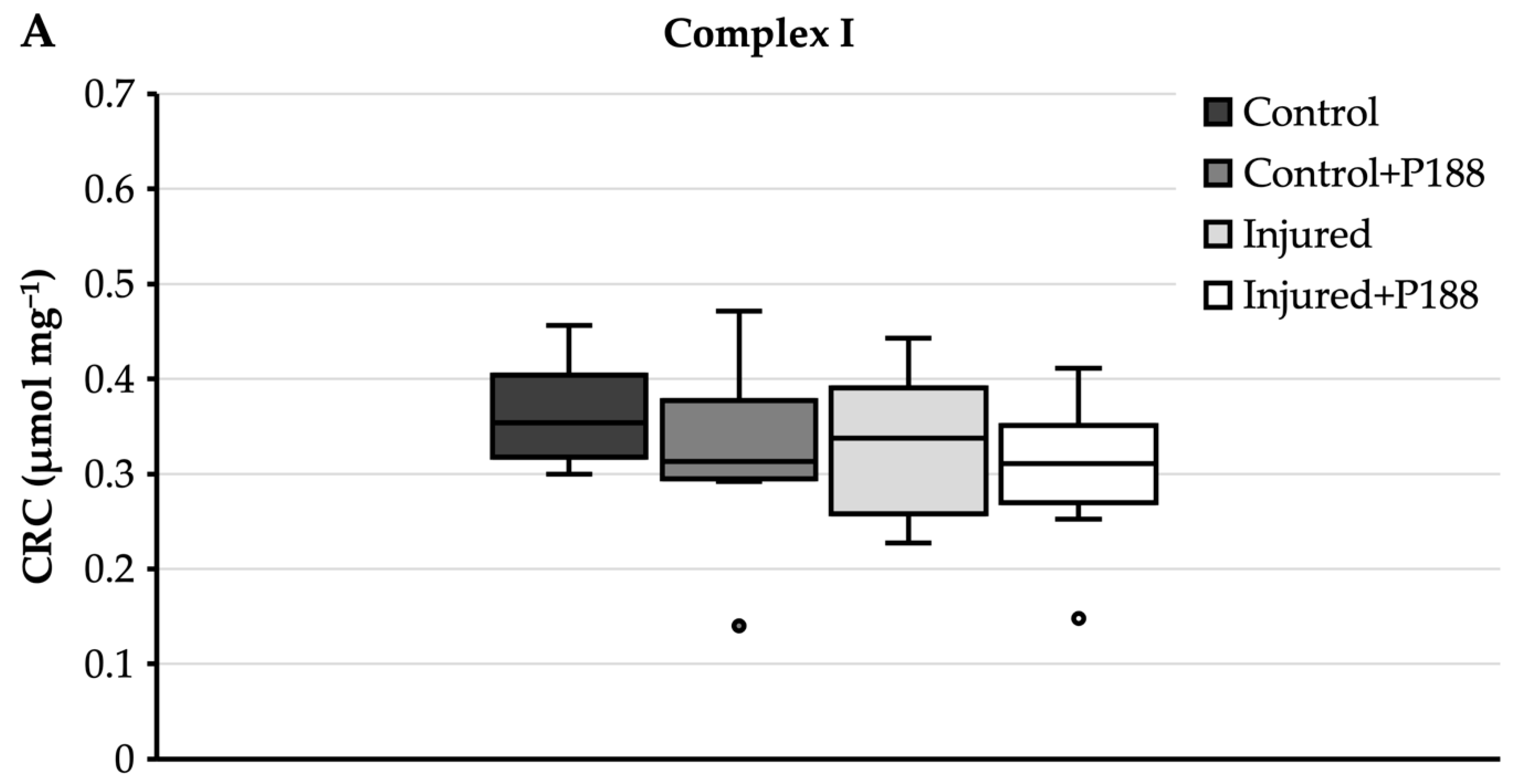
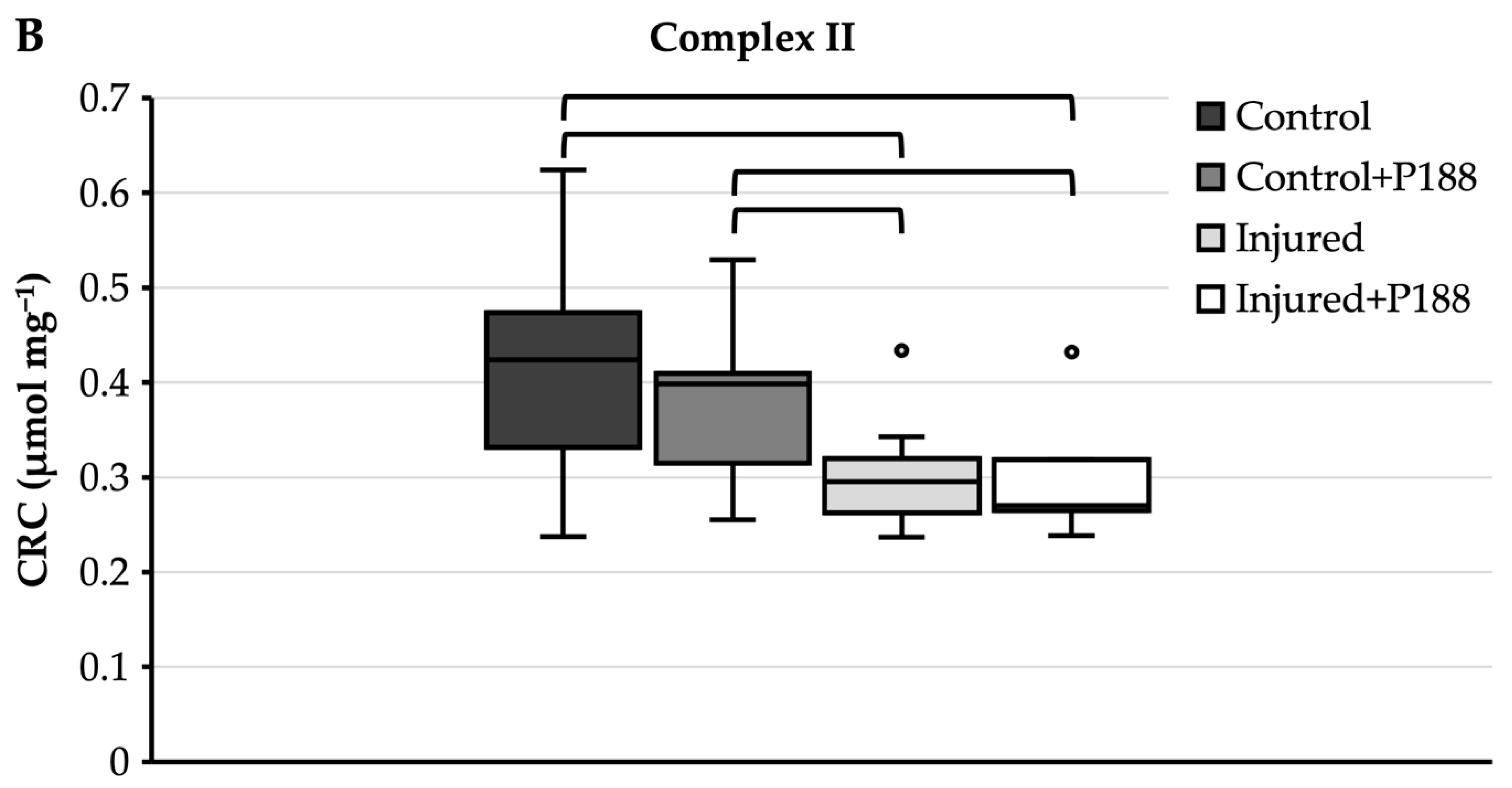
3.1.3. CRC after H2O2-Induced Injury
3.2. Comparison of Different Exposure Times to RT
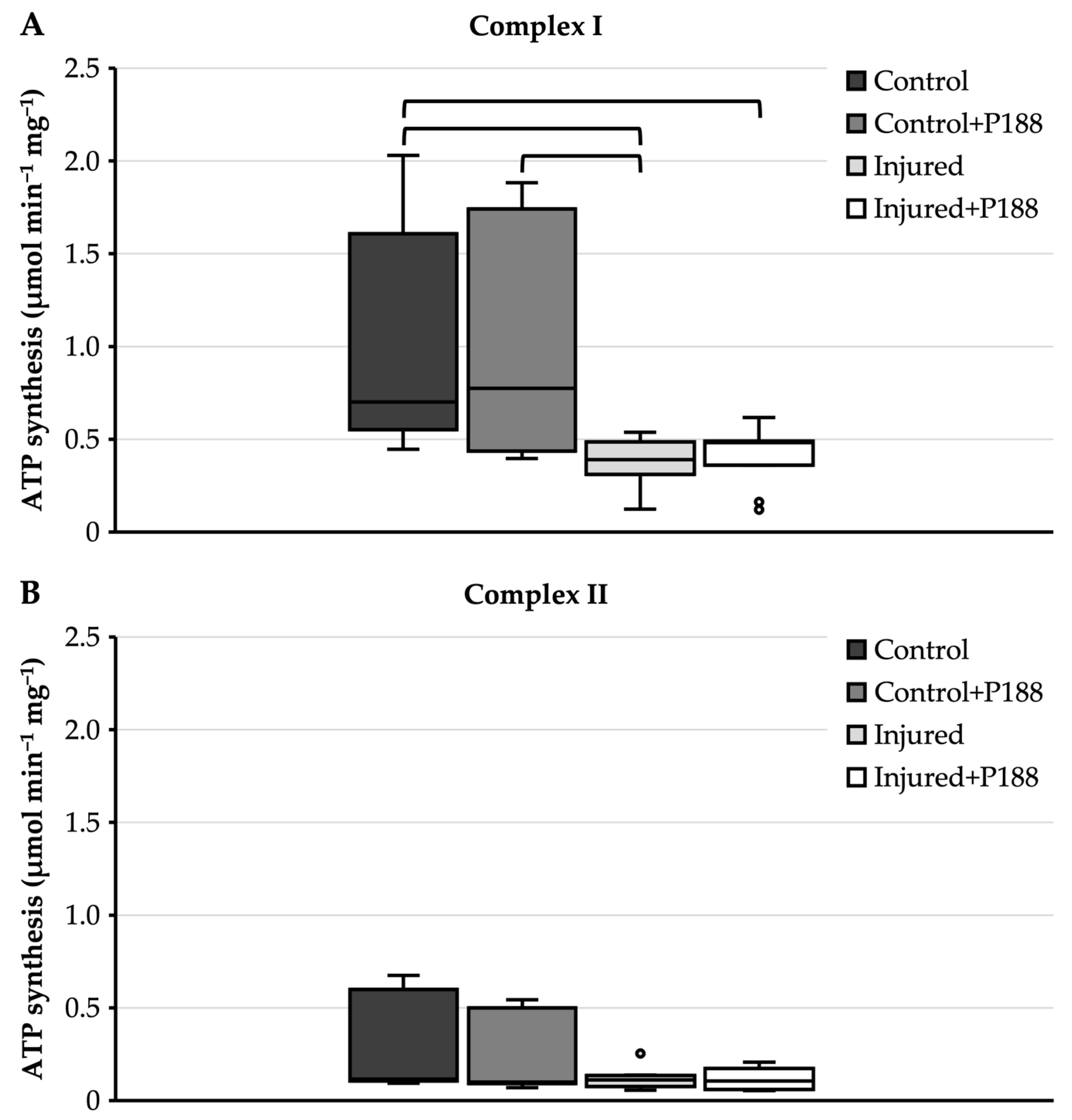
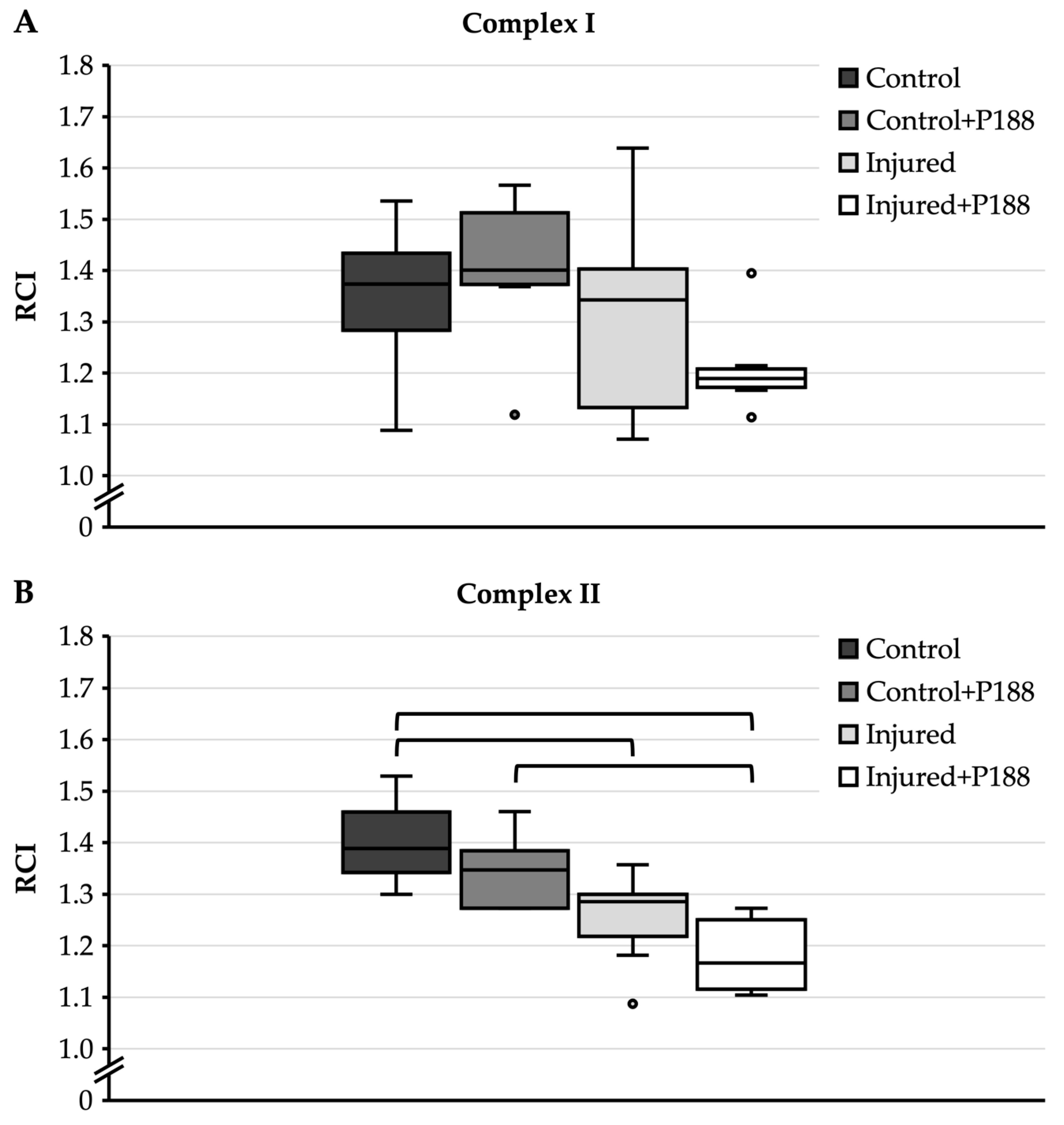
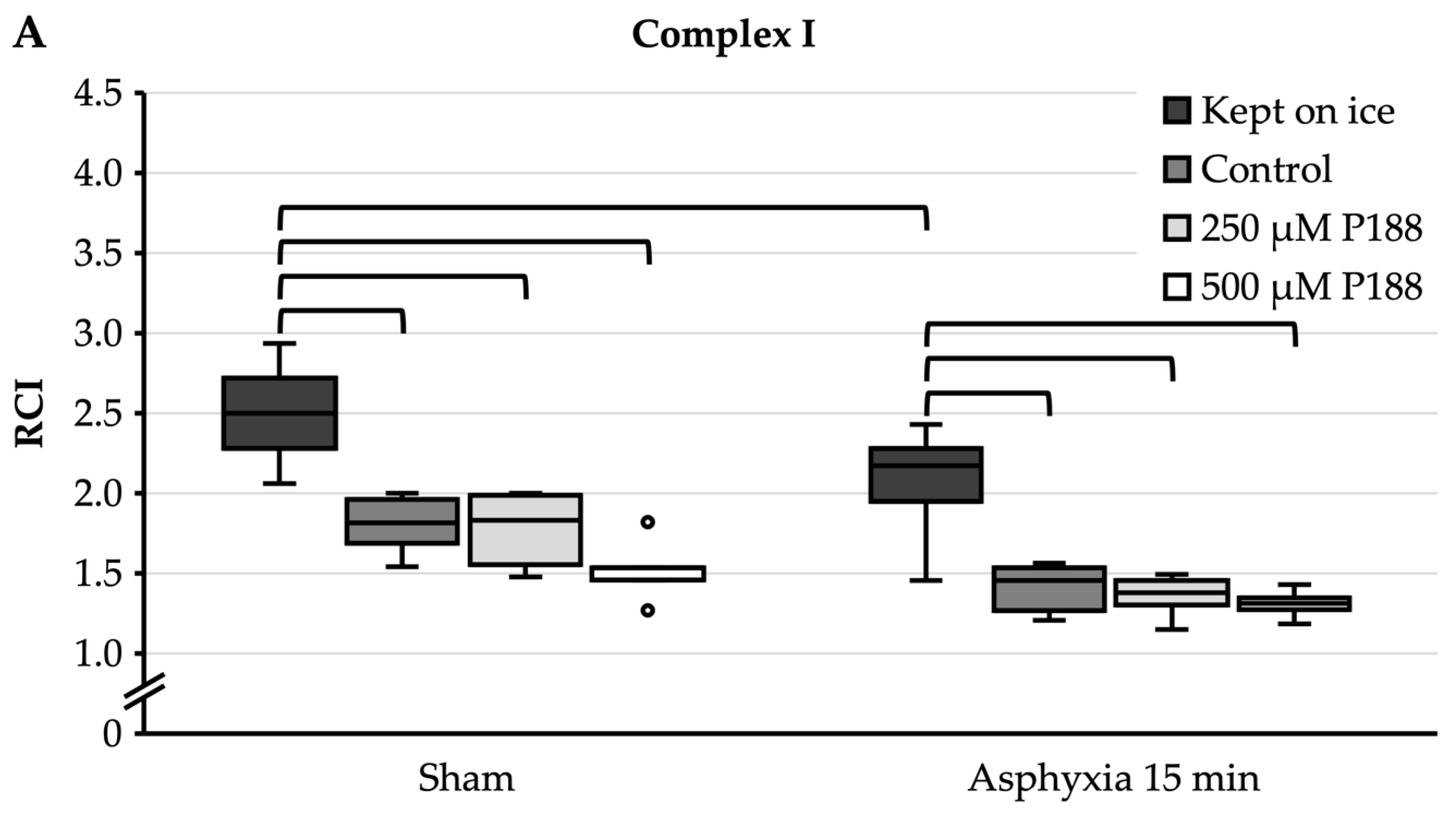
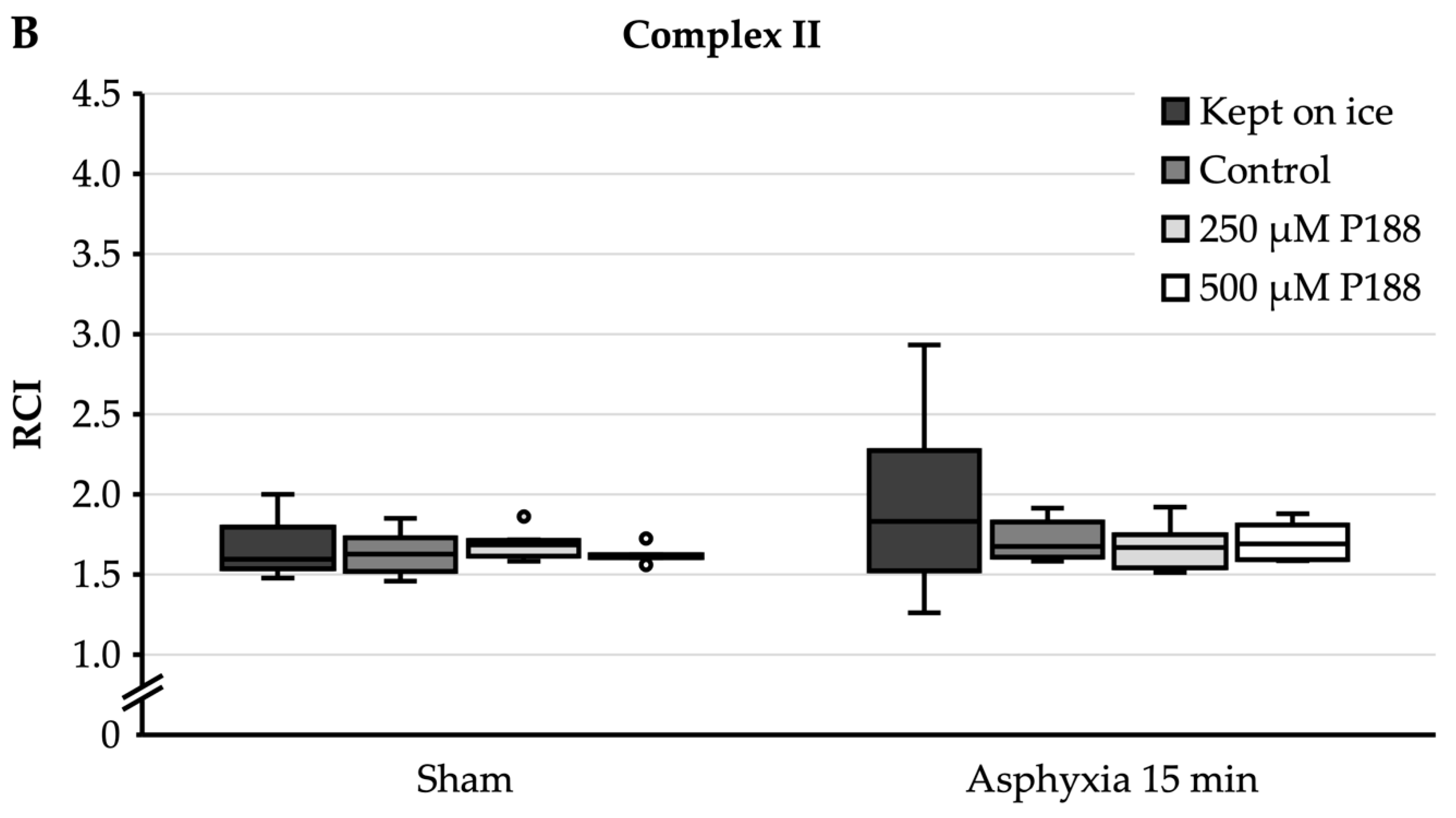
3.3. Isolated Mitochondria after Asphyxial CA-Induced Injury
4. Discussion
4.1. Methods of Injuring Isolated Mitochondria
4.1.1. In Vivo Injury
4.1.2. In Vitro Isolated Mitochondria Injury
4.2. Effect of P188 on Mitochondria
4.3. Study Limitations
- As shown above, rat isolated brain mitochondria reacted very sensibly to exposure to RT, making this alone a factor that is difficult to account for. The exposure to RT for only 10 more min significantly decreased ATP synthesis of isolated mitochondria. Similarly, in a study by Kleinbongard et al. isolated mitochondria showed lower mitochondrial respiration between a time control sample of mouse isolated heart mitochondria which was exposed to RT for 9 min and a baseline sample which was analyzed directly after isolation [44]. While no further research was found on this specific topic, these results may account for a confounding factor in many studies involving isolated mitochondria, including ours.
- When given after the isolation of mitochondria, P188 did not preserve mitochondrial function after asphyxial CA in vivo. While this concurs with the results achieved in H2O2-injured mitochondria, it is not known how well the state of mitochondria is being preserved throughout the isolation process and if the state of ischemic damage is actually “frozen” in the mitochondria. Therefore, a possible prolongation of ischemic damage inside the mitochondria could be an explanation for why P188 did not alter mitochondrial function after asphyxial CA in vivo.
- Mitochondrial function was fairly variable. This, generally, poses the question of how much mitochondria become impaired during the isolation process itself. Therefore, it is possible that mitochondria are additionally injured during the isolation such that P188 cannot rescue them anymore.
- As mentioned above, mitochondrial impairment by H2O2 occurs in a concentration-dependent manner [50]. It may very well be possible that, in the present study, mitochondria were injured too excessively and that a smaller concentration of H2O2 would have allowed for the injury to be attenuated better by P188.
- In the present study, P188 was only used on mitochondria at concentrations of or exceeding 250 µM. However, in Wang et al.’s study, the optimal concentration seemed to be at 30 µM [18]. Possibly, P188 is only able to attenuate impairment in isolated mitochondria at lower concentrations such as the ones tested in the present study.
- Lastly, the mitochondrial yield from any given animal was typically very low, so that only a few data points per animal could be extracted, making the study resource-intensive and limiting the number of animals used. Thus, insufficient power [57] and a subsequent type II error might explain why some of the results have not achieved significance despite strong trends.
4.4. Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Feigin, V.L.; Norrving, B.; Mensah, G.A. Global Burden of Stroke. Circ. Res. 2017, 120, 439–448. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients with Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [PubMed]
- Turc, G.; Bhogal, P.; Fischer, U.; Khatri, P.; Lobotesis, K.; Mazighi, M.; Schellinger, P.D.; Toni, D.; de Vries, J.; White, P.; et al. European Stroke Organisation (ESO)—European Society for Minimally Invasive Neurological Therapy (ESMINT) Guidelines on Mechanical Thrombectomy in Acute Ischaemic StrokeEndorsed by Stroke Alliance for Europe (SAFE). Eur. Stroke J. 2019, 4, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell. Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed] [Green Version]
- Madathil, R.J.; Hira, R.S.; Stoeckl, M.; Sterz, F.; Elrod, J.B.; Nichol, G. Ischemia reperfusion injury as a modifiable therapeutic target for cardioprotection or neuroprotection in patients undergoing cardiopulmonary resuscitation. Resuscitation 2016, 105, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Yu, W.; Lou, Z.; Mu, D.; Wang, Y.; Shen, B.; Qi, S. Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats. PLoS ONE 2012, 7, e46498. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartos, J.A.; Matsuura, T.R.; Tsangaris, A.; Olson, M.; McKnite, S.H.; Rees, J.N.; Haman, K.; Shekar, K.C.; Riess, M.L.; Bates, F.S.; et al. Intracoronary Poloxamer 188 Prevents Reperfusion Injury in a Porcine Model of ST-Segment Elevation Myocardial Infarction. JACC Basic Transl. Sci. 2016, 1, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Li, Q.; Gao, Y.; Shen, X.; Ma, L.; Wu, Q.; Wang, Z.; Zhang, M.; Zhao, Z.; Chen, X.; et al. Poloxamer 188 Attenuates Cerebral Hypoxia/Ischemia Injury in Parallel with Preventing Mitochondrial Membrane Permeabilization and Autophagic Activation. J. Mol. Neurosci. 2015, 56, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.L.; Chen, X.P.; Li, L.L.; Li, Q.Q.; Li, B.X.; Xue, A.M.; Xu, H.F.; Dai, D.K.; Shen, Y.W.; Tao, L.Y.; et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J. Neurotrauma 2013, 30, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D.; Gallo, G.; Barbee, K.A. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp. Neurol. 2008, 212, 422–430. [Google Scholar] [CrossRef]
- Marks, J.D.; Pan, C.Y.; Bushell, T.; Cromie, W.; Lee, R.C. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001, 15, 1107–1109. [Google Scholar] [PubMed]
- Gu, J.H.; Ge, J.B.; Li, M.; Xu, H.D.; Wu, F.; Qin, Z.H. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE 2013, 8, e61641. [Google Scholar] [CrossRef] [PubMed]
- Justicz, A.G.; Farnsworth, W.V.; Soberman, M.S.; Tuvlin, M.B.; Bonner, G.D.; Hunter, R.L.; Martino-Saltzman, D.; Sink, J.D.; Austin, G.E. Reduction of myocardial infarct size by poloxamer 188 and mannitol in a canine model. Am. Heart J. 1991, 122, 671–680. [Google Scholar] [CrossRef]
- Houang, E.M.; Bartos, J.; Hackel, B.J.; Lodge, T.P.; Yannopoulos, D.; Bates, F.S.; Metzger, J.M. Cardiac Muscle Membrane Stabilization in Myocardial Reperfusion Injury. JACC Basic Transl. Sci. 2019, 4, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Bindokas, V.P.; Skinner, M.; Emrick, T.; Marks, J.D. Mitochondrial mechanisms of neuronal rescue by F-68, a hydrophilic Pluronic block co-polymer, following acute substrate deprivation. Neurochem. Int. 2017, 109, 126–140. [Google Scholar] [CrossRef]
- Salzman, M.M.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Protects Isolated Adult Mouse Cardiomyocytes from Reoxygenation Injury. Pharmacol. Res. Perspect. 2020, 8, e00639. [Google Scholar] [CrossRef]
- Alberts, B. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2017. [Google Scholar]
- Lamoureux, L.; Radhakrishnan, J.; Gazmuri, R.J. A Rat Model of Ventricular Fibrillation and Resuscitation by Conventional Closed-chest Technique. J. Vis. Exp. 2015, e52413. [Google Scholar] [CrossRef] [Green Version]
- Kristian, T. Isolation of mitochondria from the CNS. Curr. Protoc. Neurosci. 2010, 52, 7.22.1–7.22.12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmuhamedov, E.L.; Wang, L.; Terzic, A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 1999, 519, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Katz, L.; Ebmeyer, U.; Safar, P.; Radovsky, A.; Neumar, R. Outcome model of asphyxial cardiac arrest in rats. J. Cereb. Blood Flow Metab. 1995, 15, 1032–1039. [Google Scholar] [CrossRef]
- Matsuura, T.R.; Bartos, J.A.; Tsangaris, A.; Shekar, K.C.; Olson, M.D.; Riess, M.L.; Bienengraeber, M.; Aufderheide, T.P.; Neumar, R.W.; Rees, J.N.; et al. Early Effects of Prolonged Cardiac Arrest and Ischemic Postconditioning during Cardiopulmonary Resuscitation on Cardiac and Brain Mitochondrial Function in Pigs. Resuscitation 2017, 116, 8–15. [Google Scholar] [CrossRef]
- Riess, M.L.; Camara, A.K.; Heinen, A.; Eells, J.T.; Henry, M.M.; Stowe, D.F. KATP channel openers have opposite effects on mitochondrial respiration under different energetic conditions. J. Cardiovasc. Pharmacol. 2008, 51, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Riess, M.L.; Matsuura, T.R.; Bartos, J.A.; Bienengraeber, M.; Aldakkak, M.; McKnite, S.H.; Rees, J.N.; Aufderheide, T.P.; Sarraf, M.; Neumar, R.W.; et al. Anaesthetic Postconditioning at the Initiation of CPR Improves Myocardial and Mitochondrial Function in a Pig Model of Prolonged Untreated Ventricular Fibrillation. Resuscitation 2014, 85, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.N.; Fiskum, G.; Beal, M.F. Mitochondria in neurodegeneration: Bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 1999, 19, 231–245. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 29 December 2020).
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousand Oaks, CA, USA, 2019. [Google Scholar]
- DPLYR: A Grammar of Data Manipulation. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 29 December 2020).
- GGPUBR: ‘ggplot2’ Based Publication Ready Plots. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 29 December 2020).
- Agricolae: Statistical Procedures for Agricultural Research. Available online: https://CRAN.R-project.org/package=agricolae (accessed on 29 December 2020).
- Userfriendlyscience: Quantitative Analysis Made Accessible. Available online: https://userfriendlyscience.com (accessed on 29 December 2020).
- Effects of RheothRx on mortality, morbidity, left ventricular function, and infarct size in patients with acute myocardial infarction. Collaborative Organization for RheothRx Evaluation (CORE). Circulation 1997, 96, 192–201.
- Moloughney, J.G.; Weisleder, N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat. Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef]
- Poellmann, M.J.; Lee, R.C. Repair and Regeneration of the Wounded Cell Membrane. Regen. Eng. Transl. Med. 2017, 3, 111–132. [Google Scholar] [CrossRef]
- Kilinc, D.; Gallo, G.; Barbee, K.A. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp. Neurol. 2009, 219, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelat, P.B.; Plant, L.D.; Wang, J.C.; Lee, E.; Marks, J.D. The membrane-active tri-block copolymer pluronic F-68 profoundly rescues rat hippocampal neurons from oxygen-glucose deprivation-induced death through early inhibition of apoptosis. J. Neurosci. 2013, 33, 12287–12299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lesnefsky, E.J. Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore. FEBS Lett. 2011, 585, 921–926. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Xu, H.; Xu, A.; Ross, T.; Bowler, E.; Hu, Y.; Lesnefsky, E.J. Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS ONE 2015, 10, e0118834. [Google Scholar] [CrossRef] [Green Version]
- Crestanello, J.A.; Doliba, N.M.; Babsky, A.M.; Doliba, N.M.; Niibori, K.; Osbakken, M.D.; Whitman, G.J. Opening of potassium channels protects mitochondrial function from calcium overload. J. Surg. Res. 2000, 94, 116–123. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Gedik, N.; Witting, P.; Freedman, B.; Klöcker, N.; Heusch, G. Pleiotropic, heart rate-independent cardioprotection by ivabradine. Br. J. Pharmacol. 2015, 172, 4380–4390. [Google Scholar] [CrossRef] [Green Version]
- Korge, P.; Honda, H.M.; Weiss, J.N. Protection of cardiac mitochondria by diazoxide and protein kinase C: Implications for ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2002, 99, 3312–3317. [Google Scholar] [CrossRef] [Green Version]
- Korge, P.; John, S.A.; Calmettes, G.; Weiss, J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex II. J. Biol. Chem. 2017, 292, 9896–9905. [Google Scholar] [CrossRef] [Green Version]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Malis, C.D.; Bonventre, J.V. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. A model for post-ischemic and toxic mitochondrial damage. J. Biol. Chem. 1986, 261, 14201–14208. [Google Scholar] [CrossRef]
- Ozcan, C.; Bienengraeber, M.; Dzeja, P.P.; Terzic, A. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H531–H539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, C.; Terzic, A.; Bienengraeber, M. Effective pharmacotherapy against oxidative injury: Alternative utility of an ATP-sensitive potassium channel opener. J. Cardiovasc. Pharmacol. 2007, 50, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, Y.; Nakaki, J.; Kawasaki, T.; Koda, K.; Ago, Y.; Baba, A.; Matsuda, T. Edaravone, a radical scavenger, inhibits mitochondrial permeability transition pore in rat brain. J. Pharmacol. Sci. 2007, 103, 434–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thummasorn, S.; Shinlapawittayatorn, K.; Khamseekaew, J.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Humanin directly protects cardiac mitochondria against dysfunction initiated by oxidative stress by decreasing complex I activity. Mitochondrion 2018, 38, 31–40. [Google Scholar] [CrossRef]
- Kristián, T.; Siesjö, B.K. Calcium in ischemic cell death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef]
- Kirillova, G.P.; Mokhova, E.N.; Dedukhova, V.I.; Tarakanova, A.N.; Ivanova, V.P.; Efremova, N.V.; Topchieva, I.N. The influence of pluronics and their conjugates with proteins on the rate of oxygen consumption by liver mitochondria and thymus lymphocytes. Biotechnol. Appl. Biochem. 1993, 18, 329–339. [Google Scholar]
- Rapoport, N.; Marin, A.P.; Timoshin, A.A. Effect of a polymeric surfactant on electron transport in HL-60 cells. Arch. Biochem. Biophys. 2000, 384, 100–108. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Sedgwick, P. The importance of statistical power. BMJ 2013, 347, f6282. [Google Scholar] [CrossRef] [Green Version]
- 1302 Microcathode Oxygen Electrodes. Available online: http://www.strathkelvin.com/wp-content/uploads/2015/04/electrode-manual.pdf (accessed on 13 April 2020).
- Mitocell: Instructions for Use. Available online: http://www.strathkelvin.com/wp-content/uploads/2015/04/mt200-mt200A-manual.pdf (accessed on 13 April 2020).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pille, J.A.; Riess, M.L. Potential Effects of Poloxamer 188 on Rat Isolated Brain Mitochondria after Oxidative Stress In Vivo and In Vitro. Brain Sci. 2021, 11, 122. https://doi.org/10.3390/brainsci11010122
Pille JA, Riess ML. Potential Effects of Poloxamer 188 on Rat Isolated Brain Mitochondria after Oxidative Stress In Vivo and In Vitro. Brain Sciences. 2021; 11(1):122. https://doi.org/10.3390/brainsci11010122
Chicago/Turabian StylePille, Johannes A., and Matthias L. Riess. 2021. "Potential Effects of Poloxamer 188 on Rat Isolated Brain Mitochondria after Oxidative Stress In Vivo and In Vitro" Brain Sciences 11, no. 1: 122. https://doi.org/10.3390/brainsci11010122
APA StylePille, J. A., & Riess, M. L. (2021). Potential Effects of Poloxamer 188 on Rat Isolated Brain Mitochondria after Oxidative Stress In Vivo and In Vitro. Brain Sciences, 11(1), 122. https://doi.org/10.3390/brainsci11010122