Aberrant Splicing in GJB1 and the Relevance of 5′ UTR in CMTX1 Pathogenesis

 ,
,
Abstract
:
1. Introduction
2. Patients, Materials and Methods
2.1. Mutational Analysis
2.2. Transcriptional Analysis
2.3. Immunofluorescence Study
3. Results
3.1. Patients
3.2. Mutational Analysis
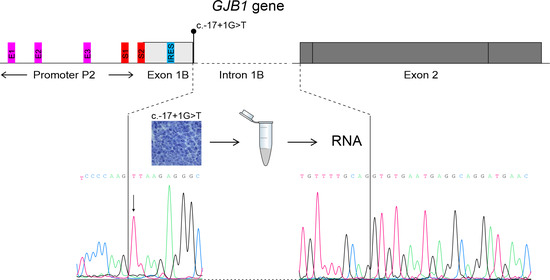
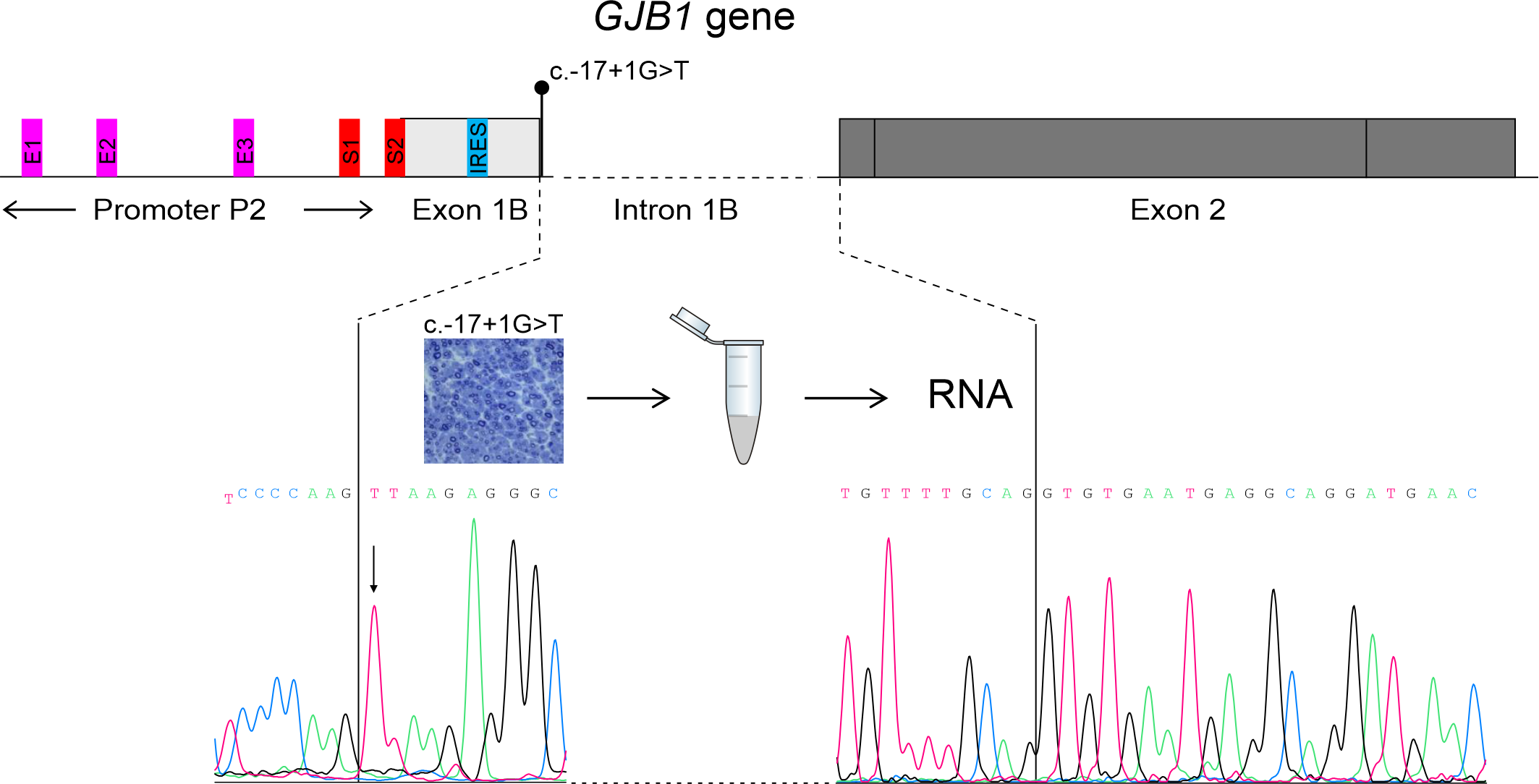
3.3. Transcriptional Analysis
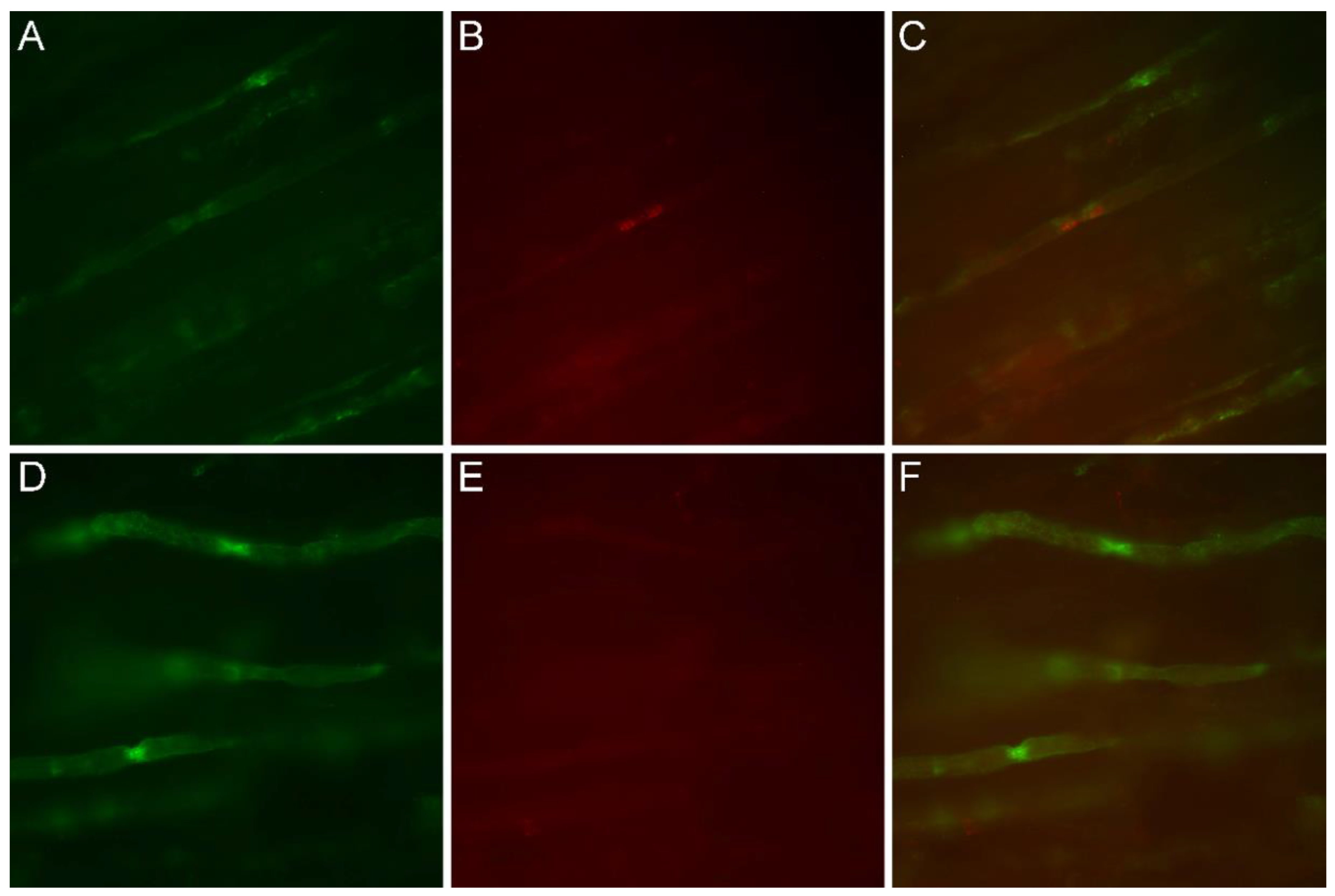
3.4. Immunofluorescence Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, S.M.; Laura, M.; Fawcett, K.; Pandraud, A.; Liu, Y.T.; Davidson, G.L.; Rossor, A.M.; Polke, J.M.; Castleman, V.; Manji, H.; et al. Charcot-Marie-Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Fridman, V.; Bundy, B.; Reilly, M.M.; Pareyson, D.; Bacon, C.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 873–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, A.; Wilcox, J.E.; Polke, J.M.; Poh, R.; Skorupinska, M.; Rossor, A.M.; Laura, M.; Tomaselli, P.J.; Houlden, H.; Shy, M.E.; et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 2020, 94, e51–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bis-Brewer, D.M.; Fazal, S.; Züchner, S. Genetic modifiers and non-Mendelian aspects of CMT. Brain Res. 2020, 1726, 146459. [Google Scholar] [CrossRef]
- Neuhaus, I.M.; Bone, L.; Wang, S.; Ionasescu, V.; Werner, R. The human connexin32 gene is transcribed from two tissue-specific promoters. Biosci. Rep. 1996, 16, 239–248. [Google Scholar] [CrossRef]
- Tomaselli, P.J.; Rossor, A.M.; Horga, A.; Jaunmuktane, Z.; Carr, A.; Saveri, P.; Piscosquito, G.; Pareyson, D.; Laura, M.; Blake, J.C.; et al. Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology 2017, 88, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Bortolozzi, M. What’s the Function of Connexin 32 in the Peripheral Nervous System? Front. Mol. Neurosci. 2018, 11, 227. [Google Scholar] [CrossRef]
- Taioli, F.; Cabrini, I.; Cavallaro, T.; Acler, M.; Fabrizi, G.M. Inherited demyelinating neuropathies with micromutations of peripheral myelin protein 22 gene. Brain 2011, 134, 608–617. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Divincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; et al. The allelic spectrum of charcot–marie–tooth disease in over 17,000 individuals with neuropathy. Mol. Genet. Genomic Med. 2014, 2, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.S.; Deschênes, S.M.; Xu, Y.T.; Grinspan, J.B.; Fischbeck, K.H.; Paul, D.L. Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 1995, 15, 8281–8294. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, G.M.; Cavallaro, T.; Taioli, F.; Polo, A.; Uncini, A.; Rizzuto, N. X-dominant Charcot-Marie-Tooth disease (CMTX): A demyelinating or axonal neuropathy? J. Peripher. Nerv. Syst. 2002, 7, 75. [Google Scholar] [CrossRef]
- Ressot, C.; Bruzzone, R. Connexin channels in Schwann cells and the development of the X-linked form of Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2000, 5, 247. [Google Scholar] [CrossRef]
- Shy, M.E.; Siskind, C.; Swan, E.R.; Krajewski, K.M.; Doherty, T.; Fuerst, D.R.; Ainsworth, P.J.; Lewis, R.A.; Scherer, S.S.; Hahn, A.F. CMT1X phenotypes represent loss of GJB1 gene function. Neurology 2007, 68, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, P.J.; Bolton, C.F.; Murphy, B.C.; Stuart, J.A.; Hahn, A.F. Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum. Genet. 1998, 103, 242–244. [Google Scholar] [CrossRef]
- Hahn, A.F.; Ainsworth, P.J.; Naus, C.C.; Mao, J.; Bolton, C.F. Clinical and pathological observations in men lacking the gap junction protein connexin 32. Muscle Nerve. Suppl. 2000, 9. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Zhang, F.; Towne, C.F.; Batish, S.D.; Lupski, J.R. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot-Marie-Tooth disease. Neurogenetics 2010, 11, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Ri, Y.; Bennett, M.V.L.; Trexler, E.B.; Verselis, V.K.; Bargiello, T.A. Changes in permeability caused by connexin 32 mutations underlie X- linked Charcot-Marie-Tooth disease. Neuron 1997, 19, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Rabadan-Diehl, C.; Dahl, G.; Werner, R. A connexin-32 mutation associated with Charcot-Marie-Tooth disease does not affect channel formation in oocytes. FEBS Lett. 1994, 351, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Deschênes, S.M.; Walcott, J.L.; Wexler, T.L.; Scherer, S.S.; Fischbeck, K.H. Altered trafficking of mutant connexin32. J. Neurosci. 1997, 17, 9077–9084. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.P.; Colman, D.R. Glial-neuron interactions and the regulation of myelin formation. Curr. Opin. Neurobiol. 1993, 5, 779–785. [Google Scholar] [CrossRef]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, post-translational modifications, and trafficking in health and disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omori, Y.; Mesnil, M.; Yamasaki, H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: Functional defects and dominant negative effects. Mol. Biol. Cell 1996, 7, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudder, A.; Werner, R. Analysis of a Charcot-Marie-Tooth disease mutation reveals an essential internal ribosome entry site element in the connexin-32 gene. J. Biol. Chem. 2000, 275, 34586–34591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionasescu, V.V.; Searby, C.; Ionasescu, R.; Neuhaus, I.M.; Werner, R. Mutations of the noncoding region of the connexin32 gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Neurology 1996, 47, 541–544. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Burton-Jones, S.; Antoniadi, T.; Rogers, M.; Jaunmuktane, Z.; Brandner, S.; Kiely, N.; Manuel, R.; Willis, T. Deletion of P2 promoter of GJB1 gene a cause of Charcot-Marie-Tooth disease. Neuromuscul. Disord. 2017, 27, 766–770. [Google Scholar] [CrossRef]
- Bondurand, N. Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 2001, 10, 2783–2795. [Google Scholar] [CrossRef] [Green Version]
- Houlden, H.; Girard, M.; Cockerell, C.; Ingram, D.; Wood, N.W.; Goossens, M.; Walker, R.W.H.; Reilly, M.M. Connexin 32 promoter P2 mutations: A mechanism of peripheral nerve dysfunction. Ann. Neurol. 2004, 56, 730–734. [Google Scholar] [CrossRef]
- Flagiello, L.; Cirigliano, V.; Strazzullo, M.; Cappa, V.; Ciccodicola, A.; D’Esposito, M.; Torrente, I.; Werner, R.; Di Iorio, G.; Rinaldi, M.; et al. Mutation in the nerve-specific 5’non-coding region of Cx32 gene and absence of specific mRNA in a CMTX1 Italian family. Mutations in brief no. 195. Online. Hum. Mutat. 1998, 12, 361. [Google Scholar]
- Benedetti, S.; Previtali, S.C.; Coviello, S.; Scarlato, M.; Cerri, F.; Di Pierri, E.; Piantoni, L.; Spiga, I.; Fazio, R.; Riva, N.; et al. Analyzing histopathological features of rare Charcot-Marie-Tooth neuropathies to unravel their pathogenesis. Arch. Neurol. 2010, 67, 1498–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.M.; Polke, J.; Manji, H.; Blake, J.; Reiniger, L.; Sweeney, M.; Houlden, H.; Brandner, S.; Reilly, M.M. A novel mutation in the nerve-specific 5′UTR of the GJB1 gene causes X-linked Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Sargiannidou, I.; Kim, G.H.; Kyriakoudi, S.; Eun, B.L.; Kleopa, K.A. A start codon CMT1X mutation associated with transient encephalomyelitis causes complete loss of Cx32. Neurogenetics 2015, 16, 193–200. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (CMTES) | Gender | Onset | Main Symptoms/Signs | Nerve Conduction Studies | Nerve Biopsy | |
|---|---|---|---|---|---|---|
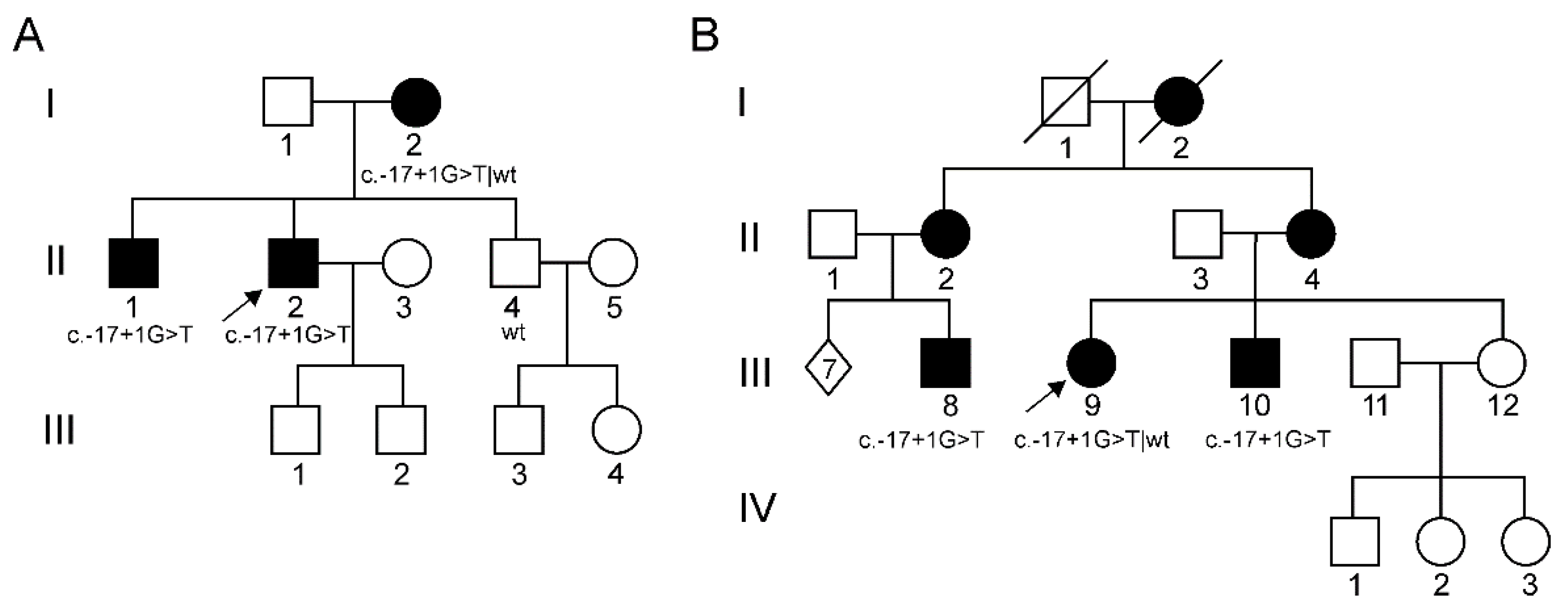
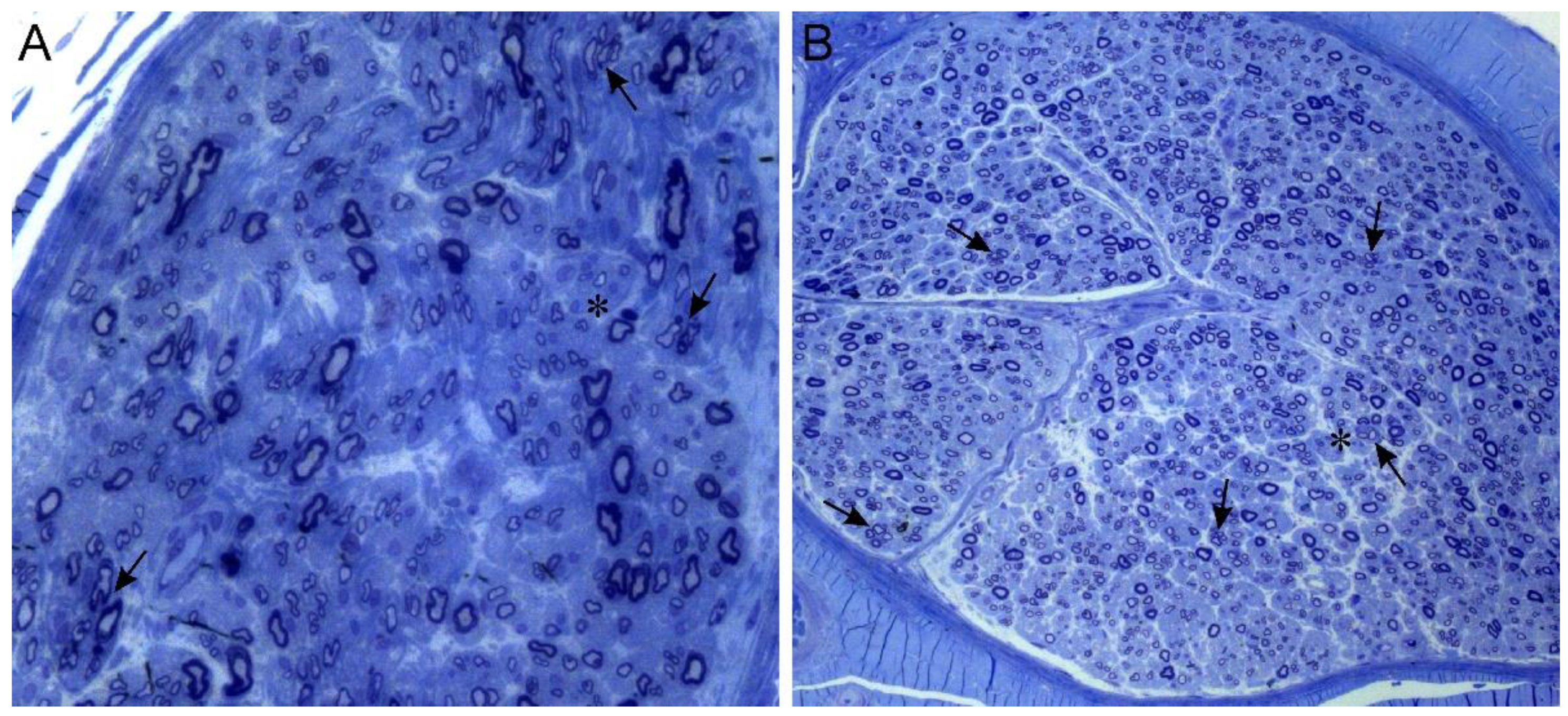
| 1st family (Figure 1A) | II-2 (12) | M | II decade (hand tremor and cramps) | 54 y.o.: Stepping gait (needing ankle-foot-orthoses) with peroneal hypotrophy and areflexia. Pes equinovarus with griffe of toes; split hand. Distal and severe hypopallesthesia with ataxic gait. Cramps at rest and during exercise. Mild postural tremor of upper limbs. Bilateral hypoacusia. | 17 and 54 y.o.: Severe reduction of cMAP of peroneal (1.5 mV → not evokable) and then median nerves (0.6 mV) with progressive decrease in conduction velocities (44 m/s and 29 m/s, respectively). Preserved sural nerve: SAP = 9 μV; SNCV = 27 m/s. | 17 y.o.: Loss of large nerve fibers, rare regeneration clusters (Figure 2A) |
| II-1 (13) | M | II decade (walking and running difficulties since he was 10) | 22 y.o.: Stepping gait with peroneal hypotrophy and weakness, lower limbs’ areflexia and distal sensory loss (feet apallesthesia) with ataxia and deficient proprioception; underwent surgery because of pes cavus at 15 years of age. Upper limbs: tremor and progressive weakness since he was 17; hyporeflexia; hand and forearm muscular hypotrophy. | |||
| I-2 (3) | F | V decade (mild walking difficulties) | 44 y.o.: Bilateral pes cavus, mild weakness in foot plantar flexion (while walking on toes) 84 y.o.: Still paucisymptomatic | |||
| 2nd family (Figure 1B) | III-9 (4) | F | IV decade (mild walking difficulties) | 33 y.o.: Pes cavus; mild weakness in hallux and foot dorsiflexion (MRC 4+/5); stocking-like sensory loss; preserved deep tendon reflexes and muscle trophism | 38 y.o.: Reduction of peroneal cMAP (2 mV); non-evokable sural SAP; intermediate motor conduction velocities (37 m/s for both peroneal and median nerves, 40 m/s for ulnar nerve). | |
| III-10 (11) | M | III decade (walking difficulties and progressive distal atrophy) | 28 y.o.: Stepping gait with ankle-foot orthosis; loss of deep tendon reflexes; pes equinovarus; “stoking and glove” deep sensory loss; ataxia. Simian hand. Upper limb postural tremor. | 15 y.o.: Axonal neuropathy, mainly affecting large fibers; signs of regeneration | ||
| III-8 (8) | M | II decade (walking difficulties) | 18 y.o.: Stepping gait with lower limbs’ distal hypotrophy and areflexia; bilateral pes cavus. Preserved strength and deep tendon reflexes on upper limbs. | 17 y.o.: Reduction of peroneal cMAP (2 mV); decrease in motor conduction velocities (32 m/s for peroneal nerve, 36 m/s for median nerve) | 18 y.o.: Axonal neuropathy with moderate reduction of large nerve fibers; sparse regeneration clusters of small fibers (Figure 2B) | |
| I-2; II-2; II-4 | F | N/A | Mild walking difficulties |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boso, F.; Taioli, F.; Cabrini, I.; Cavallaro, T.; Fabrizi, G.M. Aberrant Splicing in GJB1 and the Relevance of 5′ UTR in CMTX1 Pathogenesis. Brain Sci. 2021, 11, 24. https://doi.org/10.3390/brainsci11010024
Boso F, Taioli F, Cabrini I, Cavallaro T, Fabrizi GM. Aberrant Splicing in GJB1 and the Relevance of 5′ UTR in CMTX1 Pathogenesis. Brain Sciences. 2021; 11(1):24. https://doi.org/10.3390/brainsci11010024
Chicago/Turabian StyleBoso, Federica, Federica Taioli, Ilaria Cabrini, Tiziana Cavallaro, and Gian Maria Fabrizi. 2021. "Aberrant Splicing in GJB1 and the Relevance of 5′ UTR in CMTX1 Pathogenesis" Brain Sciences 11, no. 1: 24. https://doi.org/10.3390/brainsci11010024
APA StyleBoso, F., Taioli, F., Cabrini, I., Cavallaro, T., & Fabrizi, G. M. (2021). Aberrant Splicing in GJB1 and the Relevance of 5′ UTR in CMTX1 Pathogenesis. Brain Sciences, 11(1), 24. https://doi.org/10.3390/brainsci11010024