
Biological Pathways Associated with Neuroprogression in Bipolar Disorder


{kind=link}
{kind=link}
Abstract
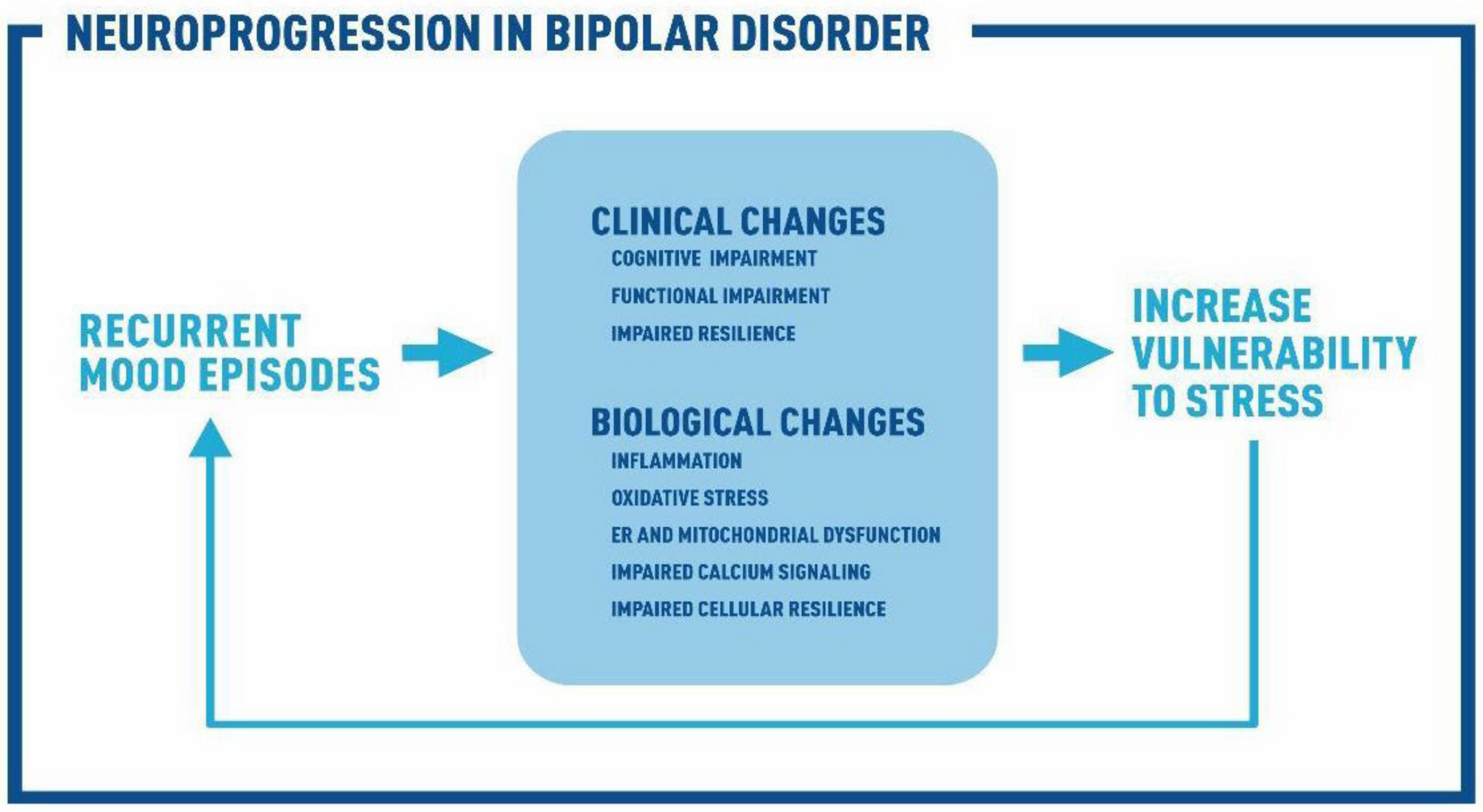
:1. Introduction
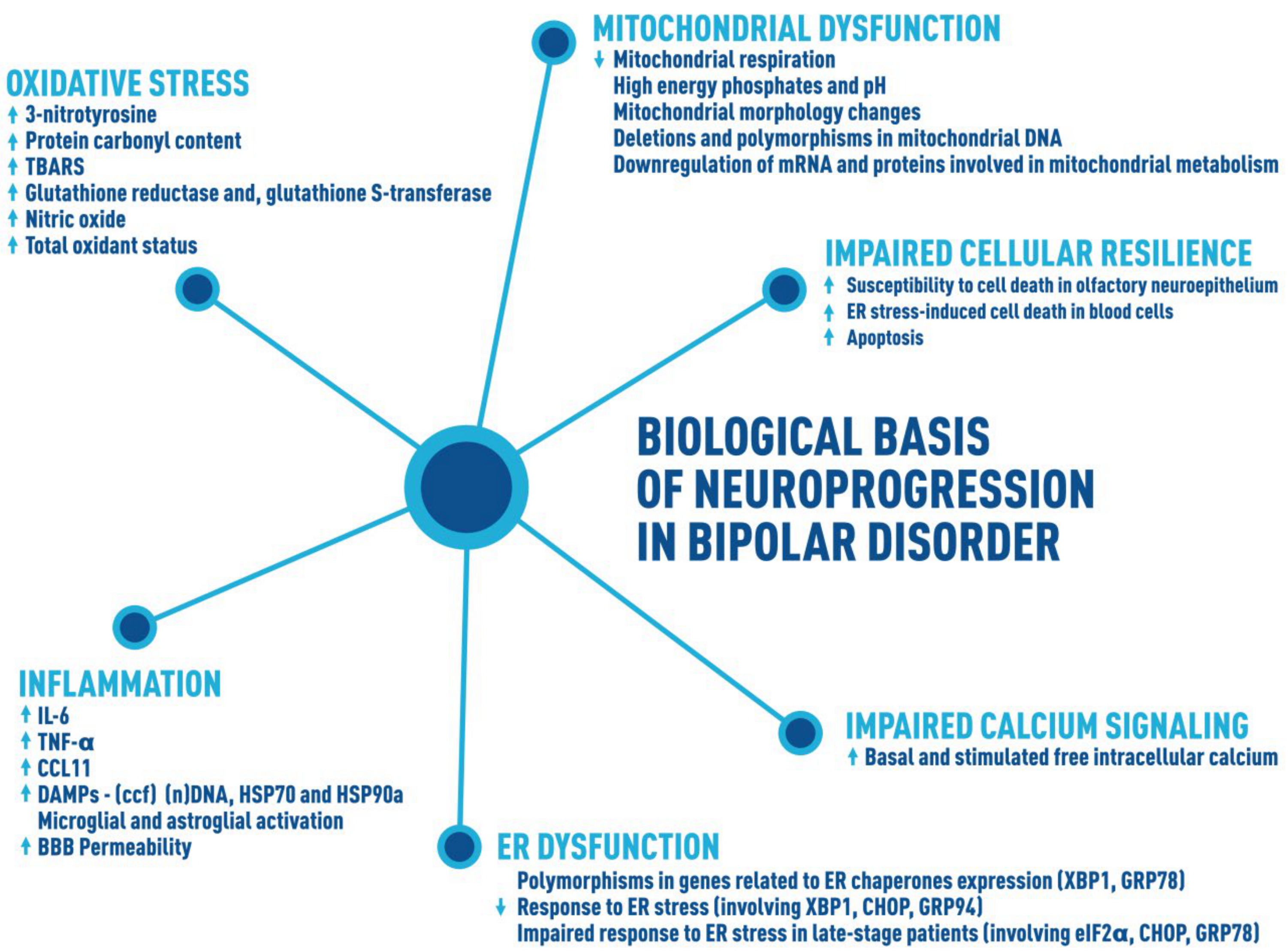
2. Inflammation
3. Impaired Cellular Resilience
4. Impaired Calcium Signaling
5. Mitochondrial Dysfunction
6. Oxidative Stress
7. Endoplasmic Reticulum Dysfunction
8. Biological Basis of Neuroprogression
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Mental Disorders. Fact Sheet N°396. Available online: www.who.int/mediacentre/factsheets/fs396/en/ (accessed on 1 October 2020).
- Kessing, L.V.; Vradi, E.; Andersen, P.K. Life expectancy in bipolar disorder. Bipolar Disord. 2015, 17, 543–548. [Google Scholar] [CrossRef]
- López-Villarreal, A.; Sánchez-Morla, E.M.; Jiménez-López, E.; Martínez-Vizcaíno, V.; Aparicio, A.I.; Mateo-Sotos, J.; Rodriguez-Jimenez, R.; Vieta, E.; Santos, J.L. Progression of the functional deficit in a group of patients with bipolar disorder: A cluster analysis based on longitudinal data. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.R.; González-Ortega, I.; González-Pinto, A.; Echeburúa, E.; Comes, M.; Martínez-Àran, A.; Ugarte, A.; Fernández, M.; Vieta, E. One-year psychosocial functioning in patients in the early vs. late stage of bipolar disorder. Acta Psychiatr. Scand. 2012, 125, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.R.; Magalhães, P.V.; Czepielewski, L.; Sulzbach, M.V.; Goi, P.D.; Vieta, E.; Gama, C.S.; Kapczinski, F. Clinical staging in bipolar disorder: Focus on cognition and functioning. J. Clin. Psychiatry 2014, 75, e450–e456. [Google Scholar] [CrossRef]
- Velosa, J.; Delgado, A.; Finger, E.; Berk, M.; Kapczinski, F.; de Azevedo Cardoso, T. Risk of dementia in bipolar disorder and the interplay of lithium: A systematic review and meta-analyses. Acta Psychiatr. Scand. 2020, 141, 510–521. [Google Scholar] [CrossRef]
- Librenza-Garcia, D.; Suh, J.S.; Watts, D.P.; Ballester, P.L.; Minuzzi, L.; Kapczinski, F.; Frey, B.N. Structural and Functional Brain Correlates of Neuroprogression in Bipolar Disorder. Curr. Top. Behav. Neurosci. 2020. Epub ahead of print. [Google Scholar]
- Cao, B.; Passos, I.C.; Mwangi, B.; Amaral-Silva, H.; Tannous, J.; Wu, M.J.; Zunta-Soares, G.B.; Soares, J.C. Hippocampal subfield volumes in mood disorders. Mol. Psychiatry. 2017, 22, 1352–1358. [Google Scholar] [CrossRef]
- Cao, B.; Passos, I.C.; Mwangi, B.; Bauer, I.E.; Zunta-Soares, G.B.; Kapczinski, F.; Soares, J.C. Hippocampal volume and verbal memory performance in late-stage bipolar disorder. J. Psychiatr. Res. 2016, 73, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, B.; Wu, M.J.; Cao, B.; Passos, I.C.; Lavagnino, L.; Keser, Z.; Zunta-Soares, G.B.; Hasan, K.M.; Kapczinski, F.; Soares, J.C. Individualized Prediction and Clinical Staging of Bipolar Disorders using Neuroanatomical Biomarkers. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 186–194. [Google Scholar] [CrossRef]
- Kessing, L.V.; Andersen, P.K. Evidence for clinical progression of unipolar and bipolar disorders. Acta Psychiatr. Scand. 2017, 135, 51–64. [Google Scholar] [CrossRef]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Bond, D.J.; Frey, B.N.; Sharma, V.; Goldstein, B.I.; Rej, S.; Beaulieu, S.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018, 20, 97–170. [Google Scholar] [CrossRef]
- Van Rheenen, T.E.; Lewandowski, K.E.; Bauer, I.E.; Kapczinski, F.; Miskowiak, K.; Burdick, K.E.; Balanzá-Martínez, V. Current understandings of the trajectory and emerging correlates of cognitive impairment in bipolar disorder: An overview of evidence. Bipolar Disord. 2020, 22, 13–27. [Google Scholar] [CrossRef]
- Frey, B.N.; Andreazza, A.C.; Houenou, J.; Jamain, S.; Goldstein, B.I.; Frye, M.A.; Leboyer, M.; Berk, M.; Malhi, G.S.; Lopez-Jaramillo, C.; et al. Biomarkers in bipolar disorder: A positional paper from the International Society for Bipolar Disorders Biomarkers Task Force. Aust. N. Z. J. Psychiatry 2013, 47, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Kauer-Sant’Anna, M.; Kapczinski, F.; Andreazza, A.C.; Bond, D.J.; Lam, R.W.; Young, L.T.; Yatham, L.N. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 447–458. [Google Scholar] [CrossRef]
- Tatay-Manteiga, A.; Balanzá-Martínez, V.; Bristot, G.; Tabarés-Seisdedos, R.; Kapczinski, F.; Cauli, O. Clinical staging and serum cytokines in bipolar patients during euthymia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 194–201. [Google Scholar] [CrossRef]
- Grande, I.; Magalhães, P.V.; Chendo, I.; Stertz, L.; Panizutti, B.; Colpo, G.D.; Rosa, A.R.; Gama, C.S.; Kapczinski, F.; Vieta, E. Staging bipolar disorder: Clinical, biochemical, and functional correlates. Acta Psychiatr. Scand. 2014, 129, 437–444. [Google Scholar] [CrossRef]
- Panizzutti, B.; Gubert, C.; Schuh, A.L.; Ferrari, P.; Bristot, G.; Fries, G.R.; Massuda, R.; Walz, J.; Rocha, N.P.; Berk, M.; et al. Increased serum levels of eotaxin/CCL11 in late-stage patients with bipolar disorder: An accelerated aging biomarker? J. Affect. Disord. 2015, 182, 64–69. [Google Scholar] [CrossRef]
- Rowland, T.; Perry, B.I.; Upthegrove, R.; Barnes, N.; Chatterjee, J.; Gallacher, D.; Marwaha, S. Neurotrophins, cytokines, oxidative stress mediators and mood state in bipolar disorder: Systematic review and meta-analyses. Br. J. Psychiatry 2018, 213, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Taslimi, S.; Brietzke, E.; Ashrafi, M. Cytokine alterations in bipolar disorder: A meta-analysis of 30 studies. Biol. Psychiatry 2013, 74, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, K.; Vinberg, M.; Vedel Kessing, L. Cytokines in bipolar disorder: A systematic review and meta-analysis. J. Affect. Disord. 2013, 144, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, K.; Vinberg, M.; Pedersen, B.K.; Poulsen, H.E.; Ekstrøm, C.T.; Kessing, L.V. A multisystem composite biomarker as a preliminary diagnostic test in bipolar disorder. Acta Psychiatr. Scand. 2019, 139, 227–236. [Google Scholar] [CrossRef]
- Wollenhaupt-Aguiar, B.; Librenza-Garcia, D.; Bristot, G.; Przybylski, L.; Stertz, L.; Kubiachi Burque, R.; Ceresér, K.M.; Spanemberg, L.; Caldieraro, M.A.; Frey, B.N.; et al. Differential biomarker signatures in unipolar and bipolar depression: A machine learning approach. Aust. N. Z. J. Psychiatry 2020, 54, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Kapczinski, F.; Andreazza, A.C.; Dean, O.M.; Giorlando, F.; Maes, M.; Yücel, M.; Gama, C.S.; Dodd, S.; Dean, B.; et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011, 35, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Bartoli, F.; Carrà, G.; Małecka, M.; Samochowiec, J.; Jarosz, K.; Banik, A.; Stańczykiewicz, B. Chemokine alterations in bipolar disorder: A systematic review and meta-analysis. Brain Behav. Immun. 2020, 88, 870–877. [Google Scholar] [PubMed]
- Kapczinski, F.; Dal-Pizzol, F.; Teixeira, A.L.; Magalhaes, P.V.; Kauer-Sant’Anna, M.; Klamt, F.; Moreira, J.C.; de Bittencourt Pasquali, M.A.; Fries, G.R.; Quevedo, J.; et al. Peripheral biomarkers and illness activity in bipolar disorder. J. Psychiatr. Res. 2011, 45, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.I.; Baune, B.T.; Bond, D.J.; Chen, P.H.; Eyler, L.; Fagiolini, A.; Gomes, F.; Hajek, T.; Hatch, J.; McElroy, S.L.; et al. Call to action regarding the vascular-bipolar link: A report from the Vascular Task Force of the International Society for Bipolar Disorders. Bipolar Disord. 2020, 22, 440–460. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Ng-Mak, D.S.; Stafkey-Mailey, D.; Farrelly, E.; Rajagopalan, K.; Loebel, A. Cardiometabolic comorbidities, readmission, and costs in schizophrenia and bipolar disorder: A real-world analysis. Ann. Gen. Psychiatry 2017, 16, 9. [Google Scholar] [CrossRef]
- Stertz, L.; Fries, G.R.; Rosa, A.R.; Kauer-Sant’anna, M.; Ferrari, P.; Paz, A.V.; Green, C.; Cunha, Â.B.; Dal-Pizzol, F.; Gottfried, C.; et al. Damage-associated molecular patterns and immune activation in bipolar disorder. Acta Psychiatr. Scand. 2015, 132, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Kapczinski, F.; Vieta, E.; Magalhães, P.V.S.; Berk, M. Neuroprogression and Staging in Bipolar Disorder, 1st ed.; Oxford University: Oxford, UK, 2015; p. 368. [Google Scholar]
- Kapczinski, F.; Berk, M.; Magalhães, P.V.S. Neuroprogression in Psychiatric; Oxford University Press: Oxford, UK, 2019; p. 272. [Google Scholar]
- Stertz, L.; Magalhães, P.V.; Kapczinski, F. Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr. Opin. Psychiatry 2013, 26, 19–26. [Google Scholar] [CrossRef]
- Rao, J.S.; Harry, G.J.; Rapoport, S.I.; Kim, H.W. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol. Psychiatry 2010, 15, 384–392. [Google Scholar] [CrossRef]
- Rajkowska, G.; Halaris, A.; Selemon, L.D. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry 2001, 49, 741–752. [Google Scholar] [CrossRef]
- Vostrikov, V.M.; Uranova, N.A.; Orlovskaya, D.D. Deficit of perineuronal oligodendrocytes in the prefrontal cortex in schizophrenia and mood disorders. Schizophr. Res. 2007, 94, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.A.; Vostrikov, V.M.; Orlovskaya, D.D.; Rachmanova, V.I. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: A study from the Stanley Neuropathology Consortium. Schizophr. Res. 2004, 67, 269–275. [Google Scholar] [CrossRef]
- Patel, J.P.; Frey, B.N. Disruption in the Blood-Brain Barrier: The Missing Link between Brain and Body Inflammation in Bipolar Disorder? Neural Plast. 2015, 2015, 708306. [Google Scholar] [CrossRef]
- Haarman, B.C.; Riemersma-Van der Lek, R.F.; de Groot, J.C.; Ruhé, H.G.; Klein, H.C.; Zandstra, T.E.; Burger, H.; Schoevers, R.A.; de Vries, E.F.; Drexhage, H.A.; et al. Neuroinflammation in bipolar disorder—A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 2014, 40, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Sehmbi, M.; Rowley, C.D.; Minuzzi, L.; Kapczinski, F.; Steiner, M.; Sassi, R.B.; Bock, N.A.; Frey, B.N. Association of intracortical myelin and cognitive function in bipolar I disorder. Acta Psychiatr. Scand. 2018, 138, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Aggio, V.; Pratesi, M.L.; Greco, G.; Furlan, R. Neuroinflammation in Bipolar Depression. Front. Psychiatry 2020, 11, 71. [Google Scholar] [CrossRef]
- Barbosa, I.G.; Bauer, M.E.; Machado-Vieira, R.; Teixeira, A.L. Cytokines in bipolar disorder: Paving the way for neuroprogression. Neural Plast. 2014, 2014, 360481. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Alamo, C. Monoaminergic neurotransmission: The history of the discovery of antidepressants from 1950s until today. Curr. Pharm. Des. 2009, 15, 1563–1586. [Google Scholar] [CrossRef]
- Wichers, M.C.; Maes, M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J. Psychiatry Neurosci. 2004, 29, 11–17. [Google Scholar] [PubMed]
- Jans, L.A.; Riedel, W.J.; Markus, C.R.; Blokland, A. Serotonergic vulnerability and depression: Assumptions, experimental evidence and implications. Mol. Psychiatry 2007, 12, 522–543. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Immune Dysfunction: Epidemiological Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Dodd, S.; Kauer-Sant’anna, M.; Malhi, G.S.; Bourin, M.; Kapczinski, F.; Norman, T. Dopamine dysregulation syndrome: Implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr. Scand. Suppl. 2007, 116, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G. Cell pathology in bipolar disorder. Bipolar Disord. 2002, 4, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Berk, M. Neuroprogression: Pathways to progressive brain changes in bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 441–445. [Google Scholar] [CrossRef]
- McCurdy, R.D.; Féron, F.; Perry, C.; Chant, D.C.; McLean, D.; Matigian, N.; Hayward, N.K.; McGrath, J.J.; Mackay-Sim, A. Cell cycle alterations in biopsied olfactory neuroepithelium in schizophrenia and bipolar I disorder using cell culture and gene expression analyses. Schizophr. Res. 2006, 82, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Fries, G.R.; Colpo, G.D.; Burque, R.K.; Bristot, G.; Ferrari, P.; Ceresér, K.M.; Rosa, A.R.; Klamt, F.; et al. Impaired endoplasmic reticulum stress response in bipolar disorder: Cellular evidence of illness progression. Int. J. Neuropsychopharmacol. 2014, 17, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Häcki, J.; Egger, L.; Monney, L.; Conus, S.; Rossé, T.; Fellay, I.; Borner, C. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 2000, 19, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Fries, G.R.; Vasconcelos-Moreno, M.P.; Gubert, C.; Santos, B.T.; da Rosa, A.L.; Eisele, B.; Sartori, J.; Pfaffenseller, B.; Kapczinski, F.; Kauer-Sant’anna, M. Early apoptosis in peripheral blood mononuclear cells from patients with bipolar disorder. J. Affect. Disord. 2014, 152–154, 474–477. [Google Scholar] [CrossRef]
- Pietruczuk, K.; Lisowska, K.A.; Grabowski, K.; Landowski, J.; Witkowski, J.M. Proliferation and apoptosis of T lymphocytes in patients with bipolar disorder. Sci. Rep. 2018, 8, 3327. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Fries, G.R.; Valvassori, S.S.; Zeni, C.P.; Zunta-Soares, G.; Berk, M.; Soares, J.C.; Quevedo, J. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl. Psychiatry 2017, 7, e1111. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Rapoport, S.I.; Rao, J.S. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Dis. 2010, 37, 596–603. [Google Scholar] [CrossRef]
- Gigante, A.D.; Young, L.T.; Yatham, L.N.; Andreazza, A.C.; Nery, F.G.; Grinberg, L.T.; Heinsen, H.; Lafer, B. Morphometric post-mortem studies in bipolar disorder: Possible association with oxidative stress and apoptosis. Int. J. Neuropsychopharmacol. 2011, 14, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L.; Murphy, J.; Thomas, M.; Rademacher, J. Abnormal intracellular calcium ion concentration in platelets and lymphocytes of bipolar patients. Am. J. Psychiatry 1992, 149, 118–120. [Google Scholar] [PubMed]
- Perova, T.; Wasserman, M.J.; Li, P.P.; Warsh, J.J. Hyperactive intracellular calcium dynamics in B lymphoblasts from patients with bipolar I disorder. Int. J. Neuropsychopharmacol. 2008, 11, 185–196. [Google Scholar] [CrossRef]
- Nurnberger, J.I., Jr.; Koller, D.L.; Jung, J.; Edenberg, H.J.; Foroud, T.; Guella, I.; Vawter, M.P.; Kelsoe, J.R.; Psychiatric Genomics Consortium Bipolar Group. Identification of pathways for bipolar disorder: A meta-analysis. JAMA Psychiatry 2014, 71, 657–664. [Google Scholar] [CrossRef]
- Jones, G.H.; Rong, C.; Shariq, A.S.; Mishra, A.; Machado-Vieira, R. Intracellular Signaling Cascades in Bipolar Disorder. Curr. Top. Behav. Neurosci. 2020. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling and psychiatric disease: Bipolar disorder and schizophrenia. Cell Tissue Res. 2014, 357, 477–492. [Google Scholar] [CrossRef]
- Heyes, S.; Pratt, W.S.; Rees, E.; Dahimene, S.; Ferron, L.; Owen, M.J.; Dolphin, A.C. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 2015, 134, 36–54. [Google Scholar] [CrossRef]
- Harrison, P.J.; Hall, N.; Mould, A.; Al-Juffali, N.; Tunbridge, E.M. Cellular calcium in bipolar disorder: Systematic review and meta-analysis. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; Green, E.K.; et al. Wellcome Trust Case Control Consortium. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar] [CrossRef]
- Ament, S.A.; Szelinger, S.; Glusman, G.; Ashworth, J.; Hou, L.; Akula, N.; Shekhtman, T.; Badner, J.A.; Brunkow, M.E.; Mauldin, D.E.; et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc. Natl. Acad. Sci. USA 2015, 112, 3576–3581. [Google Scholar] [CrossRef]
- Kataoka, M.; Matoba, N.; Sawada, T.; Kazuno, A.A.; Ishiwata, M.; Fujii, K.; Matsuo, K.; Takata, A.; Kato, T. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol. Psychiatry 2016, 21, 885–893. [Google Scholar] [CrossRef]
- Kato, T. Neurobiological basis of bipolar disorder: Mitochondrial dysfunction hypothesis and beyond. Schizophr. Res. 2017, 187, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Y.; Song, X.; Luo, H.; Sun, J.; Han, C.; Gu, X.; Li, J.; Cai, G.; Zhu, Y.; et al. Modulation of Stem Cells as Therapeutics for Severe Mental Disorders and Cognitive Impairments. Front. Psychiatry 2020, 11, 80. [Google Scholar] [CrossRef]
- Steardo, L., Jr.; Luciano, M.; Sampogna, G.; Carbone, E.A.; Caivano, V.; Di Cerbo, A.; Giallonardo, V.; Palummo, C.; Vece, A.; Del Vecchio, V.; et al. Clinical Severity and Calcium Metabolism in Patients with Bipolar Disorder. Brain Sci. 2020, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kim, H.K.; Song, I.S.; Youm, J.; Dizon, L.A.; Jeong, S.H.; Ko, T.H.; Heo, H.J.; Ko, K.S.; Rhee, B.D.; et al. Glucocorticoids and their receptors: Insights into specific roles in mitochondria. Prog. Biophys. Mol. Biol. 2013, 112, 44–54. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocrinol. 2018, 49, 72–85. [Google Scholar] [CrossRef]
- Belvederi Murri, M.; Prestia, D.; Mondelli, V.; Pariante, C.; Patti, S.; Olivieri, B.; Arzani, C.; Masotti, M.; Respino, M.; Antonioli, M.; et al. The HPA axis in bipolar disorder: Systematic review and meta-analysis. Psychoneuroendocrinology 2016, 63, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713. [Google Scholar] [CrossRef] [PubMed]
- Konradi, C.; Eaton, M.; MacDonald, M.L.; Walsh, J.; Benes, F.M.; Heckers, S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen. Psychiatry 2004, 61, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, J.F.; Tseng, M.; Young, L.T. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J. Psychiatry Neurosci. 2006, 31, 189–196. [Google Scholar] [PubMed]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen. Psychiatry. 2010, 67, 360–368. [Google Scholar] [CrossRef]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef]
- Dager, S.R.; Friedman, S.D.; Parow, A.; Demopulos, C.; Stoll, A.L.; Lyoo, I.K.; Dunner, D.L.; Renshaw, P.F. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen. Psychiatry 2004, 61, 450–458. [Google Scholar] [CrossRef]
- Yoshimi, N.; Futamura, T.; Bergen, S.E.; Iwayama, Y.; Ishima, T.; Sellgren, C.; Ekman, C.J.; Jakobsson, J.; Pålsson, E.; Kakumoto, K.; et al. Cerebrospinal fluid metabolomics identifies a key role of isocitrate dehydrogenase in bipolar disorder: Evidence in support of mitochondrial dysfunction hypothesis. Mol. Psychiatry 2016, 21, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Pfaffenseller, B.; Fries, G.R.; Wollenhaupt-Aguiar, B.; Colpo, G.D.; Stertz, L.; Panizzutti, B.; Magalhães, P.V.; Kapczinski, F. Neurotrophins, inflammation and oxidative stress as illness activity biomarkers in bipolar disorder. Expert Rev. Neurother. 2013, 13, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.; Tsung, A.; Billiar, T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic. Biol. Med. 2010, 48, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Young, A.H.; Juruena, M.F. The Neurobiology of Bipolar Disorder. Curr. Top. Behav. Neurosci. 2021. Advance online publication. [Google Scholar] [CrossRef]
- Kapczinski, F.; Dal-Pizzol, F.; Teixeira, A.L.; Magalhaes, P.V.; Kauer-Sant’Anna, M.; Klamt, F.; Pasquali, M.A.; Quevedo, J.; Gama, C.S.; Post, R. A systemic toxicity index developed to assess peripheral changes in mood episodes. Mol. Psychiatry 2010, 15, 784–786. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Kapczinski, F.; Kauer-Sant’Anna, M.; Walz, J.C.; Bond, D.J.; Gonçalves, C.A.; Young, L.T.; Yatham, L.N. 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J. Psychiatry Neurosci. 2009, 34, 263–271. [Google Scholar]
- Magalhães, P.V.; Jansen, K.; Pinheiro, R.T.; Colpo, G.D.; da Motta, L.L.; Klamt, F.; da Silva, R.A.; Kapczinski, F. Peripheral oxidative damage in early-stage mood disorders: A nested population-based case-control study. Int. J. Neuropsychopharmacol. 2012, 15, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Kauer-Sant’anna, M.; Frey, B.N.; Bond, D.J.; Kapczinski, F.; Young, L.T.; Yatham, L.N. Oxidative stress markers in bipolar disorder: A meta-analysis. J. Affect. Disord. 2008, 111, 135–144. [Google Scholar] [CrossRef]
- Kunz, M.; Gama, C.S.; Andreazza, A.C.; Salvador, M.; Ceresér, K.M.; Gomes, F.A.; Belmonte-de-Abreu, P.S.; Berk, M.; Kapczinski, F. Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in different phases of bipolar disorder and in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1677–1681. [Google Scholar] [CrossRef]
- Yumru, M.; Savas, H.A.; Kalenderoglu, A.; Bulut, M.; Celik, H.; Erel, O. Oxidative imbalance in bipolar disorder subtypes: A comparative study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1070–1074. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.F.; Young, L.T. Chronic valproate treatment increases expression of endoplasmic reticulum stress proteins in the rat cerebral cortex and hippocampus. Biol. Psychiatry 2000, 48, 658–664. [Google Scholar] [CrossRef]
- Shao, L.; Sun, X.; Xu, L.; Young, L.T.; Wang, J.F. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci. 2006, 78, 1317–1323. [Google Scholar] [CrossRef]
- Kakiuchi, C.; Ishigaki, S.; Oslowski, C.M.; Fonseca, S.G.; Kato, T.; Urano, F. Valproate, a mood stabilizer, induces WFS1 expression and modulates its interaction with ER stress protein GRP94. PLoS ONE 2009, 4, e4134. [Google Scholar] [CrossRef]
- Kakiuchi, C.; Iwamoto, K.; Ishiwata, M.; Bundo, M.; Kasahara, T.; Kusumi, I.; Tsujita, T.; Okazaki, Y.; Nanko, S.; Kunugi, H.; et al. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 2003, 35, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Ishiwata, M.; Nanko, S.; Kunugi, H.; Minabe, Y.; Nakamura, K.; Mori, N.; Fujii, K.; Umekage, T.; Tochigi, M.; et al. Functional polymorphisms of HSPA5: Possible association with bipolar disorder. Biochem. Biophys. Res. Commun. 2005, 336, 1136–1143. [Google Scholar] [CrossRef]
- So, J.; Warsh, J.J.; Li, P.P. Impaired endoplasmic reticulum stress response in B-lymphoblasts from patients with bipolar-I disorder. Biol. Psychiatry 2007, 62, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Toyota, T.; Yoshikawa, T.; Kato, T. Aberrant endoplasmic reticulum stress response in lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef]
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic reticulum-mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wollenhaupt-Aguiar, B.; Kapczinski, F.; Pfaffenseller, B. Biological Pathways Associated with Neuroprogression in Bipolar Disorder. Brain Sci. 2021, 11, 228. https://doi.org/10.3390/brainsci11020228
Wollenhaupt-Aguiar B, Kapczinski F, Pfaffenseller B. Biological Pathways Associated with Neuroprogression in Bipolar Disorder. Brain Sciences. 2021; 11(2):228. https://doi.org/10.3390/brainsci11020228
Chicago/Turabian StyleWollenhaupt-Aguiar, Bianca, Flavio Kapczinski, and Bianca Pfaffenseller. 2021. "Biological Pathways Associated with Neuroprogression in Bipolar Disorder" Brain Sciences 11, no. 2: 228. https://doi.org/10.3390/brainsci11020228
APA StyleWollenhaupt-Aguiar, B., Kapczinski, F., & Pfaffenseller, B. (2021). Biological Pathways Associated with Neuroprogression in Bipolar Disorder. Brain Sciences, 11(2), 228. https://doi.org/10.3390/brainsci11020228