
Homology Modelling, Molecular Docking and Molecular Dynamics Simulation Studies of CALMH1 against Secondary Metabolites of Bauhinia variegata to Treat Alzheimer’s Disease


 , , , ,
, , , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Protein Sequence Retrieval
2.2. Protein Secondary Structure Prediction
2.3. Protein Tertiary Structure Prediction through Template Identification
2.4. Modelling
2.5. Validation of the Structure
2.6. Energy Minimisation of the Predicted Molecule
2.7. Preparation of Ligand Molecule
2.8. Initial Docking through iGEMDOCK Software
2.9. Final Molecular Docking through AutoDock Vina and Drug Likeliness Property Analysis
2.10. Molecular Dynamics Simulations
3. Results and Discussion
3.1. Protein Sequence
3.2. Protein Secondary Structure Prediction
3.3. Template Identification
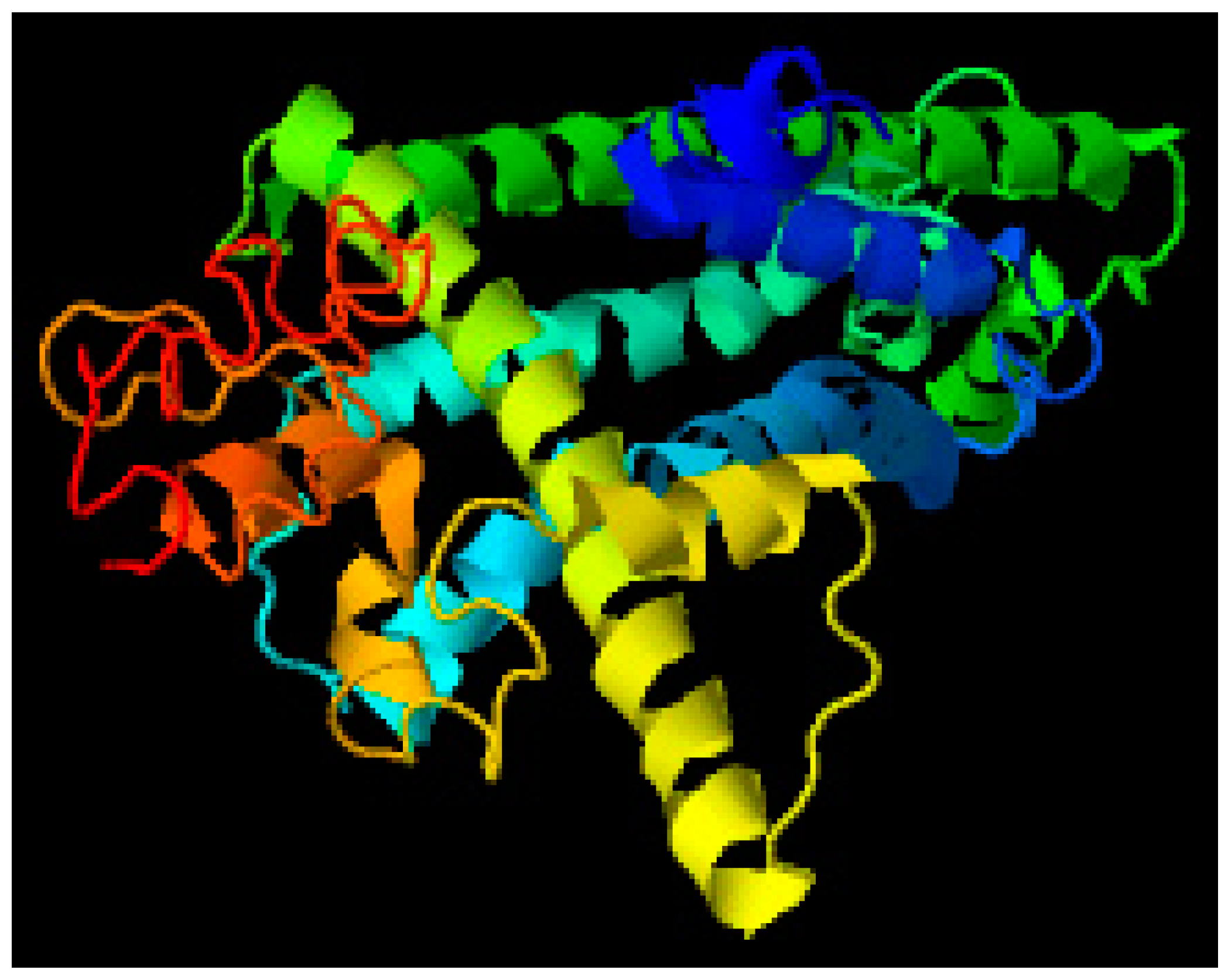
3.4. Modelling through MODELLER
3.5. Structure Prediction through LOMETS Server
3.6. Protein Structure Prediction Using MUSTER Server
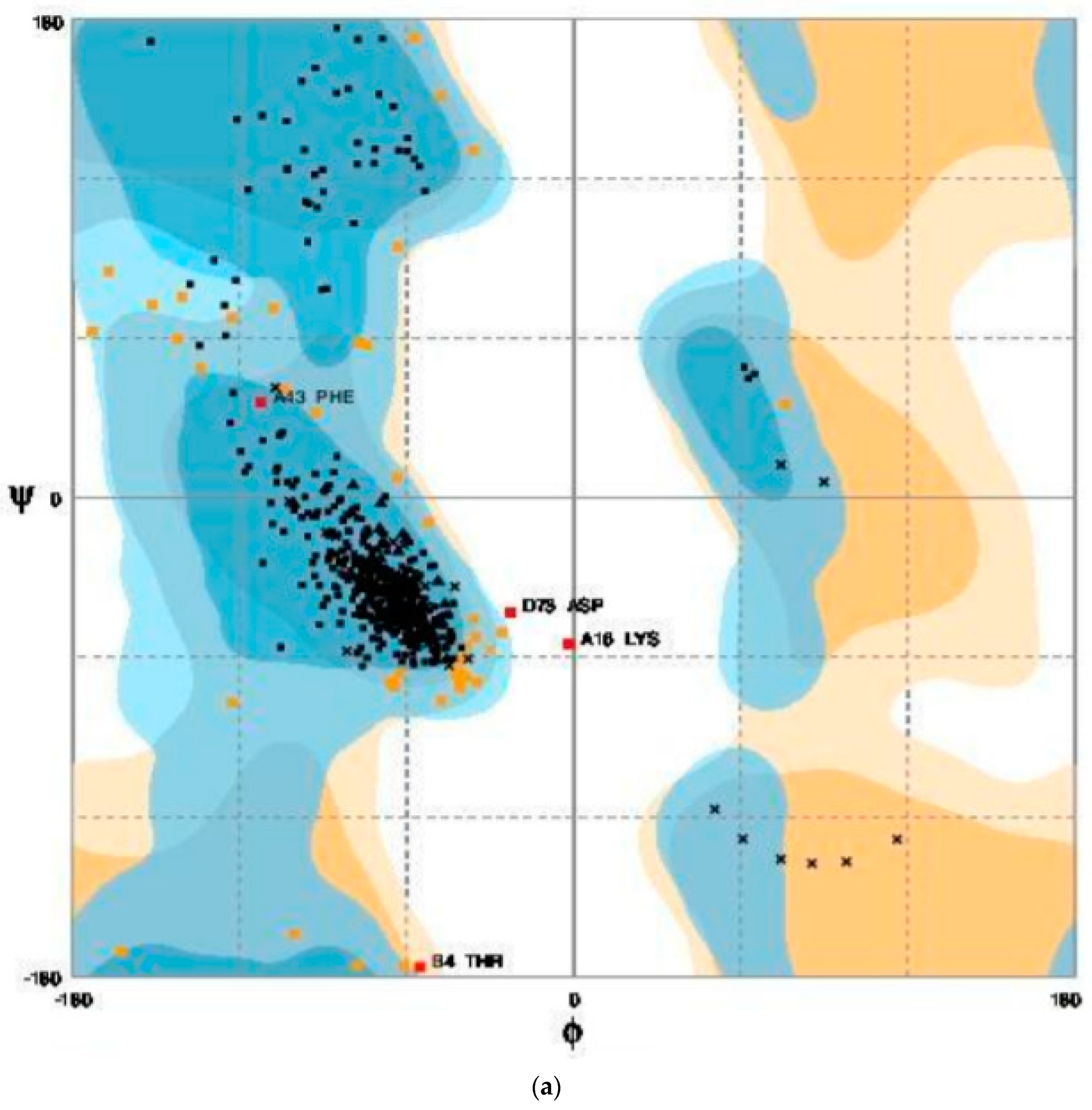
3.7. Structure Validation Using Ramachandran Plot
3.8. Energy Minimisation
3.9. Ligands from Bauhinia variegata
3.10. Screening of Ligands through iGEMDOCK
3.11. Molecular Docking Analysis through AutoDock Vina
3.12. Cheminformatics Properties and Lipinski’s Rule of Five Validation of Quercetin
3.13. Quercetin’s Pharmacokinetic Properties
3.14. Molecular Dynamic Simulations Analysis
3.15. Root Mean Square Deviation (RMSD)
3.16. Radius of Gyration
3.17. Solvent Accessible Surface Area (SASA)
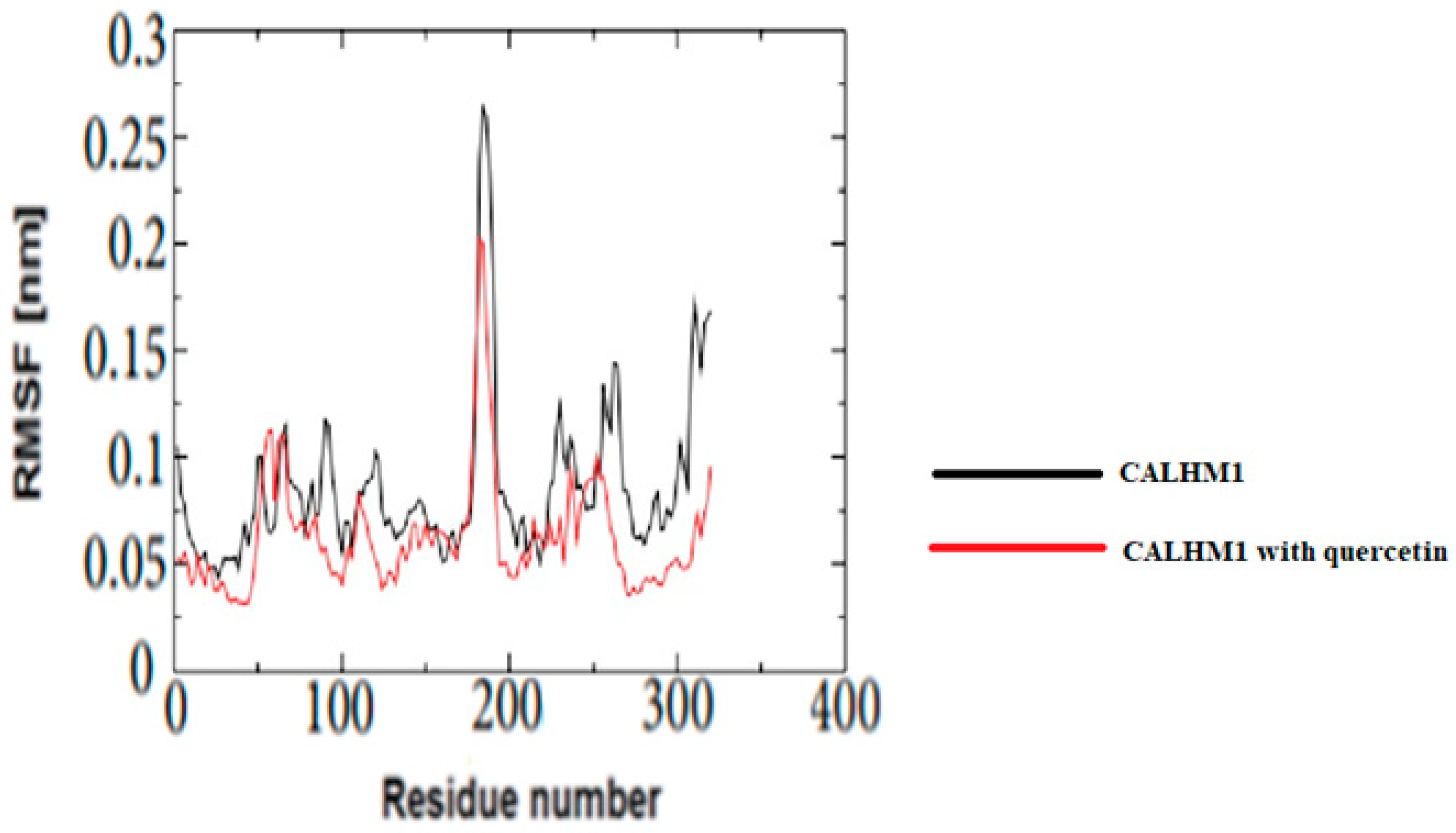
3.18. Root Mean Square Fluctuation (RMSF)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandra, P.M.; Venkateshwar, J. Biological evaluation of Schiff bases of new isatin derivatives for anti Alzheimer’s activity. Asian J. Pharm. Clin. Res. 2014, 7, 114–117. Available online: https://innovareacademics.in/journals/index.php/ajpcr/article/view/966 (accessed on 31 December 2020).
- Khare, N.; Maheshwari, S.K.; Jha, A.K. Screening and identification of secondary metabolites in the bark of Bauhinia variegata to treat Alzheimer’s disease by using molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 39, 5988–5998. [Google Scholar] [CrossRef]
- Ma, Z.; Siebert, A.P.; Cheung, K.H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, 109, E1963–E1971. [Google Scholar] [CrossRef]
- Syrjanen, J.L.; Michalski, K.; Chou, T.H.; Grant, T.; Rao, S.; Simorowski, N.; Furukawa, H. Structure and assembly of calcium homeostasis modulator proteins. Nat. Struct. Mol. Biol. 2020, 27, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.F.; Seto, S.N.; Pera, M.; Bosch, M.M.; Plata, C.; Belbin, O.; Soininen, H. Rare variants in calcium homeostasis modulator 1 (CALHM1) found in early onset Alzheimer’s disease patients alter calcium homeostasis. PLoS ONE 2013, 8, e74203. [Google Scholar]
- Ma, Z.; Tanis, J.E.; Taruno, A.; Foskett, J.K. Calcium homeostasis modulator (CALHM) ion channels. Pflüg. Arch.-Eur. J. Physiol. 2016, 468, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Koppel, J. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Nacmias, B.; Tedde, A.; Bagnoli, S.; Lucenteforte Cellini, E.; Piaceri, I.; Sorbi, S. Lack of implication for CALHM1 P86L common variation in Italian patients with early and late onset Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 37–41. [Google Scholar] [CrossRef]
- Bigiani, A. Calcium homeostasis modulator 1-like currents in rat fungiform taste cells expressing amiloride-sensitive sodium currents. Chem. Senses 2017, 42, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Dreses, W.U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Pasquier, F. A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef]
- Dreses, W.U.; Vingtdeux, V.; Zhao, H.; Chandakkar, P.; Davies, P.; Marambaud, P. CALHM1 controls the Ca2+-dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J. Cell Sci. 2013, 126, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. MUSTER: Improving protein sequence profile–profile alignments by using multiple sources of structure information. Proteins 2008, 72, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Orry, A.J.; Abagyan, R. Homology Modeling: Methods and Protocols; Humana Press: New York, NY, USA, 2012. [Google Scholar]
- Karim, R.; Aziz, M.; Al, M.; Shatabda, S.; Rahman, M.S.; Mia, M. CoMOGrad and PHOG: From computer vision to fast and accurate protein tertiary structure retrieval. Sci. Rep. 2015, 5, 13275. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Liu, J.; Wu, T.; Guo, Z.; Hou, J.; Cheng, J. Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14. Proteins 2022, 90, 58–72. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Schwede, T. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Marhefka, C.A.; Moore, B.M.; Bishop, T.C.; Kirkovsky, L.; Mukherjee, A.; Dalton, J.T.; Miller, D.D. Homology modeling using multiple molecular dynamics simulations and docking studies of the human androgen receptor ligand binding domain bound to testosterone and nonsteroidal ligands. J. Med. Chem. 2001, 44, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Khare, P.; Kishore, K.; Sharma, D. A study on the standardization parameters of Bauhinia variegate. Asian J. Pharm. Clin. Res. 2017, 10, 133–136. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Humana Press: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Nousheen, L.; Akkiraju, P.C.; Enaganti, S. Molecular docking mutational studies on human surfactant protein-D. World J. Pharm. Res. 2014, 3, 1140–1148. [Google Scholar]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graphbased signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Van, S.P.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Iqbal, D.; Khan, M.S.; Waiz, M.; Rehman, M.T.; Alaidarous, M.; Jamal, A.; Alothaim, A.S.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; et al. Exploring the Binding Pattern of Geraniol with Acetylcholinesterase through In Silico Docking, Molecular Dynamics Simulation, and In Vitro Enzyme Inhibition Kinetics Studies. Cells 2021, 10, 3533. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef] [PubMed]
- Ongey, E.L.; Yassi, H.; Pflugmacher, S.; Neubauer, P. Pharmacological and pharmacokinetic properties of lanthipeptides undergoing clinical studies. Biotechnol. Lett. 2017, 39, 473–482. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query Cover | E-Value | Identity | Accession |
|---|---|---|---|
| 99% | 6 × 10−169 | 68.36% | 6VAM A |
| 88% | 4 × 10−139 | 58.82% | 6LMT A |
| MODELLER | LOMETS | MUSTER | ||
|---|---|---|---|---|
| 6VAM A | 6LMT A | |||
| Favoured region | 187 | 172 | 177 | 196 |
| Allowed region | 5 | 6 | 14 | 10 |
| Outlier region | 2 | 3 | 15 | 6 |
| MODELLER | LOMETS | MUSTER | ||
|---|---|---|---|---|
| 6VAM A | 6LMT A | |||
| Energy (KJ/mol) | 2468.876 | 5688.255 | 10,265.889 | 8714.236 |
| Compound | PubChem ID |
|---|---|
| Hentriacontane | CID: 12410 |
| Octacosanol | CID:68406 |
| Stigmasterol | CID:5280794 |
| Betasitosterol | CID:222284 |
| Flavanone | CID:10251 |
| Isoquericetroside | CID:5484006 |
| Kaempeferol-3-glucoside | CID:6325460 |
| Lupeol | CID:259846 |
| Myricetol | CID:5281672 |
| Phenanthriquinone | CID:6763 |
| Quercitroside | CID:5280459 |
| Rutoside | CID:5280805 |
| Xanthophyll | CID:5281243 |
| Beta- carotene | CID:5280489 |
| Dihydroquercetin | CID:439533 |
| Quercetin | CID:5280343 |
| Ligands | Binding Energy | VDW | HBond |
|---|---|---|---|
| Quercetin (CID: 5280343) | −12.66 | −22.13 | −2.34 |
| Dihydroquercetin (CID: 439533) | −10.30 | −21.11 | −2.18 |
| Beta-carotene (CID: 5280489) | −10.26 | −20.11 | −3.42 |
| Xanthophylls (CID: 5281243) | −8.20 | −11.33 | −4.57 |
| Stigma sterol (CID: 5280794) | −7.80 | −29.20 | −7.6 |
| Beta-sitosterol (CID: 222284) | −6.70 | −30.29 | −3.41 |
| S.No | Mode | Affinity (kcal/mol) | Distance From Best Mode RMSD l.b | Distance From Best Mode RMSD u.b |
|---|---|---|---|---|
| 1. | 1 | −12.45 | 0.000 | 0.000 |
| 2. | 2 | −12.34 | 21.115 | 20.357 |
| 3. | 3 | −12.12 | 12.235 | 12.514 |
| 4. | 4 | −11.65 | 9.656 | 6.524 |
| 5. | 5 | −11.25 | 8.459 | 2.722 |
| 6. | 6 | −10.81 | 8.287 | 10.650 |
| 7. | 7 | −9.45 | 7.775 | 1.089 |
| 8. | 8 | −9.32 | 6.002 | 11.924 |
| 9. | 9 | −8.23 | 6.028 | 14.615 |
| Molecular Formula | C15H10O7 |
|---|---|
| Molecular weight (g/mol) | 302.24 |
| Hydrogen bond acceptor | 7 |
| Hydrogen bond donor | 5 |
| Rotatable bonds | 1 |
| Log p | 0.56 |
| No of atoms | 22 |
| Polar surface area (A2) | 103.49 A2 |
| Molar refractivity (cm3) | 122.60 |
| Density (cm3) | 1.23 |
| Molar volume (cm3) | 268.73 cm3 |
| Drug likeness | 1 |
| Lipinski validation | yes |
| GPCR ligand | −0.06 |
| Ion channel modulator | −0.19 |
| Kinase inhibitor | 0.28 |
| Nuclear receptor ligand | 0.36 |
| Protease inhibitor | −0.25 |
| Enzyme inhibitor | 0.28 |
| S.No. | Property | Model Name | Predicted Value | Unit |
|---|---|---|---|---|
| 1. | Absorption | Water solubility | −2.925 | Numeric (log mol/L) |
| 2. | Absorption | Caco2 permeability | −0.229 | Numeric (log Papp in 10–6 cm/s) |
| 3. | Absorption | Intestinal absorption (human) | 96.902 | Numeric (% absorbed) |
| 4. | Absorption | Skin permeability | −2.735 | Numeric (log Kp) |
| 5. | Absorption | P-glycoprotein substrate | Yes | Categorical (Yes/No) |
| 6. | Absorption | P-glycoprotein I inhibitor | No | Categorical (Yes/No) |
| 7. | Absorption | P-glycoprotein II inhibitor | No | Categorical (Yes/No) |
| 8. | Distribution | VDss (human) | 1.559 | Numeric (log L/kg) |
| 9. | Distribution | Fraction unbound (human) | 0.206 | Numeric (Fu) |
| 10. | Distribution | BBB permeability | −1.098 | Numeric (log BB) |
| 11. | Distribution | CNS permeability | −3.065 | Numeric (log PS) |
| 12. | Metabolism | CYP2D6 substrate | No | Categorical (yes/no) |
| 13. | Metabolism | CYP3A4 substrate | No | Categorical (yes/no) |
| 14. | Metabolism | CYP1A2 inhibitor | Yes | Categorical (yes/no) |
| 15. | Metabolism | CYP2C19 inhibitor | No | Categorical (yes/no) |
| 16. | Metabolism | CYP2C9 inhibitor | No | Categorical (yes/no) |
| 17. | Metabolism | CYP2D6 inhibitor | No | Categorical (yes/no) |
| 18. | Metabolism | CYP3A4 inhibitor | No | Categorical (yes/no) |
| 19. | Excretion | Total clearance | 0.407 | Numeric (log ml/min/kg) |
| 20. | Excretion | Renal OCT2 substrate | No | Categorical (yes/no) |
| 21. | Toxicity | AMES toxicity | No | Categorical (yes/no) |
| 22. | Toxicity | Max. tolerated dose (human) | 0.499 | Numeric (log mg/kg/day) |
| 23. | Toxicity | hERG I inhibitor | No | Categorical (yes/no) |
| 24. | Toxicity | hERG II inhibitor | No | Categorical (yes/no) |
| 25. | Toxicity | Oral rat acute toxicity (LD50) | 2.471 | Numeric (mol/kg) |
| 26. | Toxicity | Oral rat chronic toxicity (LOAEL) | 2.612 | Numeric (log mg/kg_bw/day) |
| 27. | Toxicity | Hepatotoxicity | No | Categorical (yes/no) |
| 28. | Toxicity | Skin sensitisation | No | Categorical (yes/no) |
| 29. | Toxicity | T. pyriformis toxicity | 0.288 | Numeric (log μg/L) |
| 30. | Toxicity | Minnow toxicity | 3.721 | Numeric (log mM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khare, N.; Maheshwari, S.K.; Rizvi, S.M.D.; Albadrani, H.M.; Alsagaby, S.A.; Alturaiki, W.; Iqbal, D.; Zia, Q.; Villa, C.; Jha, S.K.; et al. Homology Modelling, Molecular Docking and Molecular Dynamics Simulation Studies of CALMH1 against Secondary Metabolites of Bauhinia variegata to Treat Alzheimer’s Disease. Brain Sci. 2022, 12, 770. https://doi.org/10.3390/brainsci12060770
Khare N, Maheshwari SK, Rizvi SMD, Albadrani HM, Alsagaby SA, Alturaiki W, Iqbal D, Zia Q, Villa C, Jha SK, et al. Homology Modelling, Molecular Docking and Molecular Dynamics Simulation Studies of CALMH1 against Secondary Metabolites of Bauhinia variegata to Treat Alzheimer’s Disease. Brain Sciences. 2022; 12(6):770. https://doi.org/10.3390/brainsci12060770
Chicago/Turabian StyleKhare, Noopur, Sanjiv Kumar Maheshwari, Syed Mohd Danish Rizvi, Hind Muteb Albadrani, Suliman A. Alsagaby, Wael Alturaiki, Danish Iqbal, Qamar Zia, Chiara Villa, Saurabh Kumar Jha, and et al. 2022. "Homology Modelling, Molecular Docking and Molecular Dynamics Simulation Studies of CALMH1 against Secondary Metabolites of Bauhinia variegata to Treat Alzheimer’s Disease" Brain Sciences 12, no. 6: 770. https://doi.org/10.3390/brainsci12060770
APA StyleKhare, N., Maheshwari, S. K., Rizvi, S. M. D., Albadrani, H. M., Alsagaby, S. A., Alturaiki, W., Iqbal, D., Zia, Q., Villa, C., Jha, S. K., Jha, N. K., & Jha, A. K. (2022). Homology Modelling, Molecular Docking and Molecular Dynamics Simulation Studies of CALMH1 against Secondary Metabolites of Bauhinia variegata to Treat Alzheimer’s Disease. Brain Sciences, 12(6), 770. https://doi.org/10.3390/brainsci12060770